Key points
- •
The fact that up to 50% of atopic dermatitis (AD) patients have loss-of-function genetic mutations of skin barrier protein filaggrin points to a cutaneous weakness that allows external factors to trigger skin inflammation in AD.
- •
Various studies documented that between 30% and 100% of patients with AD have pathogenic Staphylococcus aureus colonization on their skin, and this colonization worsens skin inflammation. Powerful evidences point to the colonization of S. aureus , paralleling a dysbiosis phenomenon with displacement of commensal bacteria and reduction of skin surface microbial diversity, as a contributing factor for AD development.
- •
Patients with AD are prone to have frequent skin infections.
- •
Patients with AD, particularly those with severe disease, tend to have heightened skin sensitivity to various contact allergens and are more likely to develop contact allergic reactions.
- •
Environmental pollutions are associated with the development of AD.
Introduction
When the loss-of-function filaggrin gene mutation was first revealed in the medical literature as a strong link to patients affected with atopic dermatitis (AD), the general sentiment of the medical community was that of an excitement for possessing a key to point to external factors as the cause of AD since the skin barrier defect would naturally open doors for invading actors to enter and trigger the inflammatory process manifested in AD ( ). Combining with the clinical evidences that AD patients commonly have pathogenic bacterial colonization in their skin and frequent skin infections with bacterial, fungal, and viral microorganisms, there are strong data to support external causes for the development of AD. The principal goal of this chapter is therefore to examine and analyze those relevant clinical data to delineate the degree these external factors are contributing to the disease development. In discussing the external factors as the contributors for AD development, we must at the same time reason if external factors alone will be sufficient to cause the disease to develop.
Although clinical data are generally not considered as robust as those collected through laboratory investigations, it nevertheless provides a supporting documentation from a different angle. In a way, clinical evidence provides a sense of reality or a sense of living proof. Any medical theory or purely laboratory-based research result unsupported by clinical evidence will be called into question of whether the theory is correct or that data are relevant for the actual disease.
First, we define what is clinical evidence. According to the online 2019 Oxford Dictionary , the term clinical relates “to the observation and treatment of actual patients rather than theoretical or laboratory studies.” Another dictionary defines clinical as “(of a disease or condition) causing observable and recognizable symptoms.” Merriam-Webster Dictionary defines this way: “of, relating to, or conducted in or as if in a clinic: such as a: involving direct observation of the patient or b: based on or characterized by observable and diagnosable symptoms.” Clinical evidence therefore will be the collected data gathered from the clinical observations or studies.
Thus, to collect clinical evidence, we gather all the data relating to the symptoms and signs of what we can observe, obtain, measure about and from actual patient encounter, rather than by theoretic consideration, speculation, or purely laboratory investigation. However, laboratory data are also part of the supporting evaluation of clinical data and of documenting clinical evidence. One simple example is the clinical evaluation of early clinical failure of treatment of a gram-negative bacteria sepsis (bloodstream infection). To collect clinical evidence accurately and correctly, the clinical investigators first establish that all these patients indeed have bloodstream infection by gram-negative bacteria documented by results of blood culture, a laboratory method. Moreover, the clinical researchers have to measure many parameters to develop a set of criteria for determining the “predictors” for early clinical failure. These parameters would include some “purely clinical data” such as blood pressure, respiratory rate, altered mental status, but also include other “laboratory data” such as white blood cell count. Together, these collected data provide the valuable predictors for early clinical failure on treatment for gram-negative sepsis ( ). Similarly, to collect clinical data and to document clinical evidence in relation to development of AD, some laboratory-generated information, such as bacterial culture determinations, skin histopathology findings, genetic mutation information in skin barrier protein, immunologic status, and other clinically supportive laboratory data, is also included. The following discussions delineate clinical findings to suggest that external factors play a significant role in AD development.
Staphylococcus aureus colonization
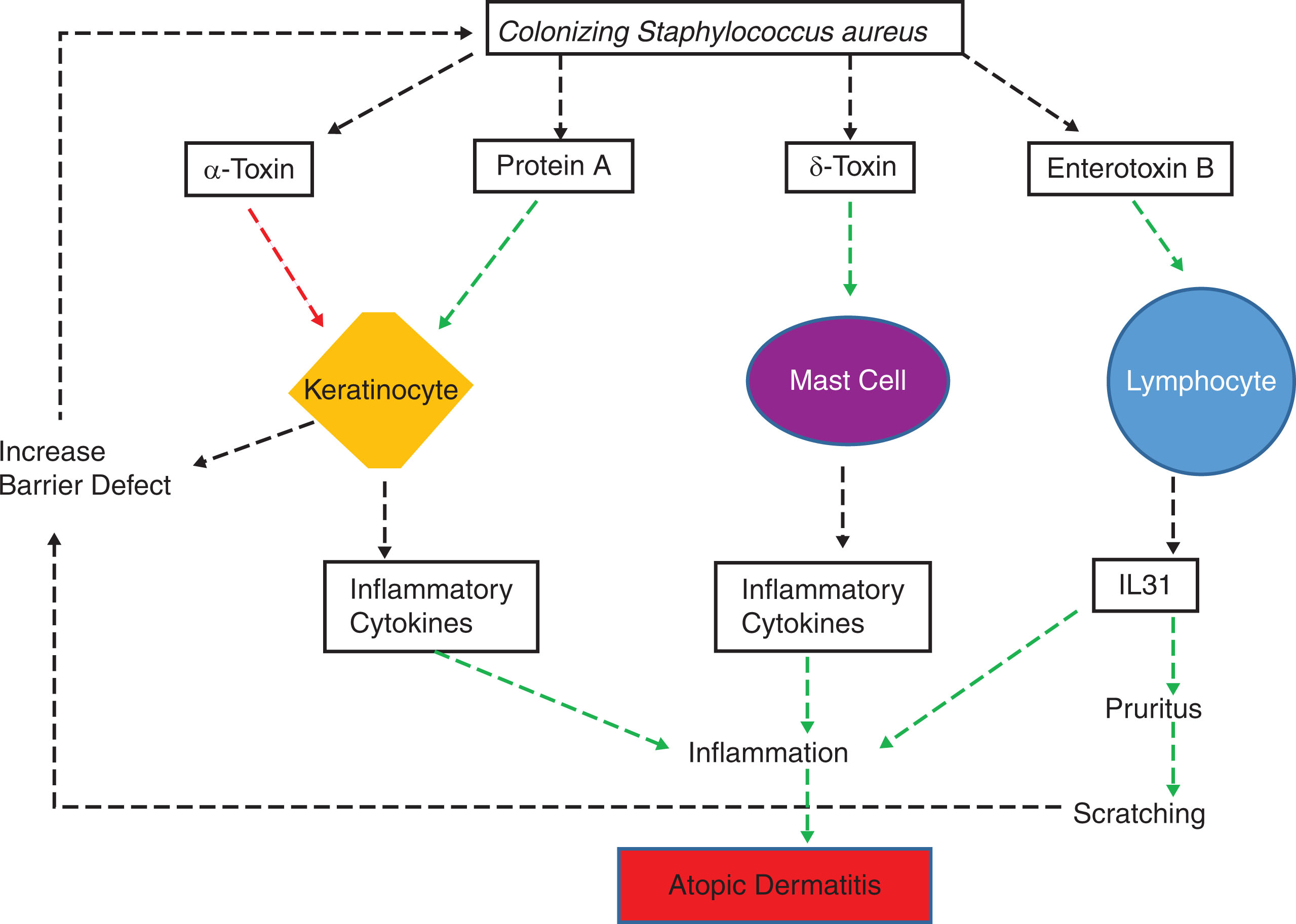
Pathogenic bacterial species colonized in the skin of AD patients is a well-documented finding. Pathogenic Staphylococcus aureus species colonize in about 30% to 100% of AD patients’ skin. This kind of colonization displaced other commensal bacteria species beneficial to the human hosts, and this abnormal colonization is associated with increased disease severity ( ). Theoretically, several molecules expressed by S. aureus may contribute to the inflammation of AD:
- •
δ-toxin from these bacteria is capable of stimulating mast cells leading to inflammatory cytokine release.
- •
α-toxin from these bacteria can damage keratinocytes leading to damage-related inflammatory responses.
- •
Phenol-soluble modulins are stimulatory to keratinocytes for cytokine release.
- •
Protein A can trigger keratinocyte inflammatory responses and act as superantigens to generate B-lymphocyte immune activation ( ).
- •
Enterotoxin B from S. aureus is capable of triggering leukocyte expression of a pruritus-inducing cytokine interleukin 31 (IL31), and experimental overexpression of IL31 in mice actually triggers skin inflammation ( ).
In one study, bacterial culture survey points out that a S. aureus– predominant colonization is associated with a severe disease phenotype, and a Staphylococcus epidermidis– predominant colonization is linked to a milder disease phenotype of AD ( ). But a mere association of S. aureus colonization does not prove a contributing role of these bacteria in triggering the disease development. One of the best studies that links S. aureus colonization to AD development is a Switzerland-conducted prospective birth cohort study. In this study of 149 white, fully term healthy infants, the researchers obtained bacterial culture at birth and at seven time points over their first 2 years of life. These infants were examined and followed by the physicians to monitor the development of skin disease at birth; at age day 1, 3, and 7; and at age 1, 3, 6, 12, and 24 months. At age 3 months, the investigators found that S. aureus colonization was significantly more prevalent in those patients who developed AD later on, compared to those infants who did not develop the skin disease. Furthermore, the prevalence of colonization increased 2 months before the onset of AD and at the time of disease onset. In addition, the patients who had positive S. aureus colonization developed AD at a younger age than those patients without the bacterial colonization ( ). There were additional negative effects of S. aureus colonization. These colonized bacteria not only settled in the skin but also pushed away the commensal skin bacteria such as S. epidermidis , which normally provides their own antimicrobial peptides and enhances human-producing antimicrobial peptides for immune defenses of human skin ( ). A proposed mechanism of how S. aureus colonization would trigger AD development is depicted in Fig. 10.1 . More discussions on the role of S. aureus in AD are detailed in Chapter 6 .
Skin infection
With the discovery of skin barrier defect, a logical reasoning to follow might be to ask if such defect would lead to a tendency of frequent skin infection in patients affected by AD. Indeed, AD has been associated with the serious bacterial, fungal, and viral infections. Eczema herpeticum and eczema vaccinatum are two such well-documented examples. However, these infections are also linked to a deficiency of innate immune defense in patients with AD, therefore raising a question of the relative contribution between internal and external actors ( ; ; ; ). Having stated the fact that frequent skin infections do occur in patients with AD, how these infections would then contribute to the development of AD is not clear. Obviously these infection events could certainly exacerbate the existing skin inflammation, but whether these infections directly trigger the skin inflammation has not been well studied.
Allergic contact dermatitis
When skin barrier function is compromised, allergens exposed to the skin may have an easy entry into the epidermis to induce allergic contact dermatitis. Once contact dermatitis, an inflammatory process by itself, begins, the development of AD could then be triggered. So the two logical questions to ask include: Are patients with AD prone to have more frequent occurrence of allergic contact dermatitis? and Do these contact allergic events lead to the typical inflammatory skin disease AD? Although the data are conflicting, existing medical publications as a whole seem to support a notion that contact allergy is a common problem in children with AD ( ). In one study, the survey results suggested that AD developed in early childhood might associate with adolescent onset of contact allergy to fragrances. However, the results of this study did not point to allergen as a causative factor for AD development, only a simple association ( ). In another study, researchers have found that patients with AD were more likely to develop positive patch test reactions to ingredients in personal care products and topically applied antibiotics and corticosteroids ( ). A study focused on difficult-to-treat AD found that this subset of patients indeed had a high prevalence of concomitant allergic contact dermatitis and were commonly sensitized to multiple allergens (polysensitized) ( ). In a Danish children study, investigators found that 30% of children with AD had at least one positive patch test reaction, and 17% of these patients had at least one contact allergic reaction relevant to their current skin symptoms, and that the risk of contact allergy significantly correlated with the AD severity. In addition, the fact that metal and skincare products were the most frequently identified allergens in this Danish study also pointed to the potential role of the external factors ( ). In another study of Dutch patients, children with AD had significantly more frequent allergic reactions to lanolin alcohol and fragrances, although the overall sensitization prevalence between atopic and nonatopic children was similar ( ). Whether these allergic contact dermatitis events directly trigger the development of AD is not well documented. An inflammatory event of allergic contact dermatitis could certainly exacerbate AD. In addition, the similar clinical morphology between AD and allergic contact dermatitis renders efforts to establish this explicit link very challenging. The association of AD with allergic contact dermatitis is also an immunologic puzzle since their disease mechanisms differ from an immune standpoint. Classically defined, AD is primarily a type 2-axis immune disease, and allergic contact dermatitis is a classic type 1 cell-mediated immune phenomenon ( ). Recent studies, however, pointed to possible common pathways, such as skin barrier defect, between allergic contact dermatitis and AD ( ). More details on the role of allergens in AD are discussed in Chapter 7 .
Environmental pollution
The discovery of skin barrier defect in patients with AD also raises the question whether the environmental pollution, airborne or by other means, may affect the development of AD. It is reasonable to speculate that when skin barrier function is deficient, the environmental pollutants could enter the skin easier. Once they enter the skin, these pollutants could in effect become irritants/allergens from an immunologic standpoint, inducing skin inflammation that we observe as AD. There were some evidences pointed to this possible link. A study conducted in South Korea found associations between development of AD and road density in patients’ home communities and between development of AD and road proximity to patients’ homes, suggesting that traffic-related air pollution positively affected the development of AD ( ). In another South Korean survey, smoking was associated with development of AD in patients age 12 to 18 years, suggesting a link between the fume of cigarette smoke and AD ( ). The fact that the incidence of AD in developing countries is slowly catching up with that of developed nations underlines the importance of environmental factors, such as hygiene practice, bacterial endotoxin exposure, animal contact, diet and intestinal microbiota, and atmospheric pollution, in the AD disease onset ( ). Thus urbanization with its associated environmental pollution could be a factor for increased prevalence of AD. To be sure, developing a link between environmental pollutions and AD with certainty is very difficult since the pollutant exposure is a chronic event, and one rarely sees the immediate skin inflammation after one such exposure. More details on the contributions of pollution in the development of AD are discussed in Chapter 9 .
Evidence from atopic march
One interesting clinical manifestation of AD is the development of atopic march, which reports the development of noncutaneous atopic diseases such as food allergy, allergic rhinitis, and asthma preceded by the cutaneous atopic disease AD. Although some evidence suggests an internal immune deviation, other evidence points to the external exposure as an important influence factor. A report on findings from the recent workshop on “atopic dermatitis and the atopic march: mechanisms and interventions” conducted by the National Institute of Allergy and Infectious Diseases provides some insights. Exposure to family dogs at birth was inversely associated with development of AD and other allergy sensitization by age 1 year and recurrent wheezing by age 3 years. Moreover, a growing body of evidence has pointed to cutaneous exposure to environmental peanut allergen (through skin barrier defect) as a leader for food allergy development. In addition, patients with AD that progresses to food allergy, especially peanut allergy, has substantial increased risk of developing airway allergic disease. Accordingly, the recommendations from this workshop support investigations of the effectiveness of early prophylactic treatments of emollients or other skin barrier enhancers for the prevention of AD and atopic march ( ). Readers are encouraged to examine evidence pointing to internal immune dysregulation as a contributing factor for AD development in Chapter 18 .
Summary
To summarize, there are many external factors identified in association with the development of AD or as potential contributing components for the development of AD. Among them are the colonization of pathogenic bacteria, other skin infections, allergens, and environmental pollutions. The big question is whether external factors alone are sufficient to induce AD or whether internal immune milieu alterations are also required to get the disease fired up. This question is germane since nearly all people are exposed to these external factors but only a fraction develop the AD disease. As of today, powerful evidences linking external factors to development of AD are limited to the colonization of the pathogenic species S. aureus . In Chapter 18 we will examine clinical evidences pointing to internal triggering factors that contribute to the development of AD.
Further readings
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


