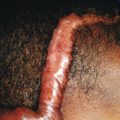
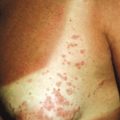
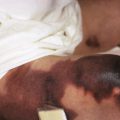
Blisters or bullae are rounded or irregularly shaped lesions of the skin or mucous membranes that result from the accumulation of fluid between the cells of the epidermis, the epidermis and stratum corneum, or the epidermis and dermis. The term bullae refers to blistering lesions 0.5 to 1 cm in diameter or larger; those smaller than 0.5 cm in diameter are called vesicles . The classification of bullous or vesiculobullous disorders is based on clinical morphology and examination of biopsied specimens of lesional or perilesional skin by light microscopy, immunofluorescence analysis, and electron microscopy. It is well recognized that the skin of infants and children is more susceptible to blister formation than that of adults.
Hereditary Blistering Disorders
Epidermolysis Bullosa
The term epidermolysis bullosa (EB) refers to a group of inherited disorders characterized by bullous lesions that develop spontaneously or as a result of varying degrees of friction or trauma. The various subtypes of inherited EB result from mutations in 18 different genes and have recently been reclassified. These phenotypes are divided into four major inherited forms based first on the level of skin cleavage ( Fig. 13-1 ) (the traditional subtypes of EB: simplex, junctional, dystrophic, and Kindler syndrome, a mixed EB type). Subsequent classification is based on the clinical phenotype including distribution of lesions and presence of scarring, the relative severity of cutaneous and extracutaneous involvement, the mode of inheritance, and if available, molecular analysis. In the new classification, the only eponyms that remain are Kindler syndrome and EB simplex (EBS)-Ogna (for lack of better suggested names).
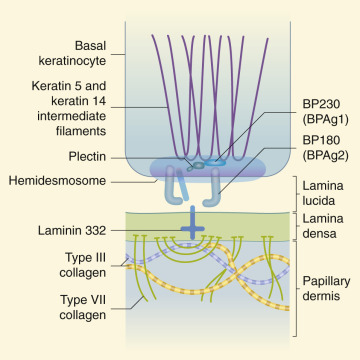
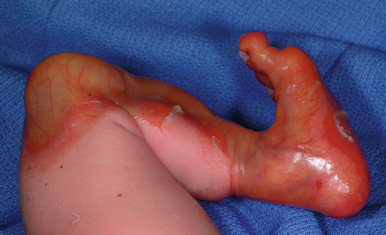
In EBS (epidermolytic EB) the blister cleavage occurs within the epidermis, and healing occurs without scarring. In junctional EB (JEB) the skin separates in the lamina lucida of the dermal–epidermal junction, and blistering leads to atrophic scarring. In dystrophic (dermolytic) EB (DEB) the blister forms in the papillary dermis below the basement membrane, and patients form scars and milia. EB with congenital absence of skin (formerly called Bart syndrome ) can be seen at birth with any of the major forms of EB ( Fig. 13-2 ), although it most commonly is seen with dystrophic forms of EB. Neonates most commonly show congenital localized absence of skin of the lower extremities. EB acquisita (EBA) is an acquired immune-mediated blistering disorder that can resemble DEB (see Epidermolysis Bullosa Acquisita section).
Skin biopsy samples for immunofluorescence mapping have traditionally been used to confirm the diagnosis of EB and to determine the subtype based on the level of cleavage from localization of known antigens in the skin and the presence of structural proteins associated with EB. Biopsies are ideally performed at the edge of a lesion freshly induced by rotating the skin with a moist Q-tip. Light microscopic evaluation of biopsy sections is generally not useful except for rare types (such as lethal acantholytic EB). Although whole exome sequencing has been shown to be more cost effective and efficient for detecting EB gene mutations than sequencing of individual genes suspected to be mutated, at this time immunomapping studies can still be performed much more quickly, providing the family with some prognostic information.
The overall incidence of EB is 1 in every 50,000 births, although the simplex forms comprise the majority of cases. A national EB registry was established in the United States and has generated data beneficial to many families who have children with EB. The Dystrophic Epidermolysis Bullosa Research Association (DEBRA), a national ( www.debra.org ) and international ( www.debra-international.org ) group, is dedicated to research and support for patients with all forms of EB and their families.
Epidermolysis Bullosa Simplex
EBS is characterized by blisters that develop in areas of trauma and most commonly results from defective keratin filaments. Because the blister cleavage is intraepidermal, lesions tend to heal without scarring. The most common form is basal EBS, which includes localized EBS (formerly called Weber–Cockayne disease ), generalized intermediate EBS (formerly called Koebner type ), and generalized severe EBS (formerly called Dowling–Meara EBS or EBS herpetiformis ) ( Table 13-1 ). The vast majority of patients with EBS show autosomal dominant inheritance, although rare autosomal recessive forms have been described. Suprabasal EBS includes a variety of autosomal recessive forms with absence of desmosomal components (for example, desmoplakin [ DSP ] in lethal acantholytic EB).
| Type | Inheritance | Affected Gene |
|---|---|---|
| EBS, suprabasal, acantholytic | AR | DSP /desmoplakin |
| EBS, suprabasal, acantholytic | AR | JUP /plakoglobin |
| EBS, suprabasal, skin fragility—plakoglobin deficiency | AR | JUP /plakoglobin |
| EBS, suprabasal, skin fragility—woolly hair | AR | DSP /desmoplakin |
| EBS, suprabasal, skin fragility—ectodermal dysplasia | AR | PKP1 /plakophilin-1 |
| EBS, suprabasal, acral peeling skin syndrome | AR | TGM5 (see Ch. 5 ) |
| Transglutaminase 5 | ||
| EBS, suprabasal; EBS superficialis | AD or AR | ? |
| EBS, basal, generalized severe (formerly Dowling–Meara) | Usually AD | KRT5, KRT14 /keratins 5, 14 |
| EBS, basal, generalized intermediate (formerly Koebner) | Usually AD | KRT5, KRT14 /keratins 5, 14 |
| EBS, basal, localized (formerly Weber–Cockayne) | Usually AD | KRT5, KRT14 /keratins 5, 14 |
| EBS, basal, with mottled pigmentation | AD | KRT5 > KRT14 |
| EBS, basal, migratory circinate | AD | KRT5 , C-terminal |
| EBS, basal, Ogna type | AD | PLEC1 /plectin |
| EBS, basal, with muscular dystrophy | AR | PLEC1 /plectin |
| EBS, basal, with pyloric atresia | AR | PLEC1 /plectin |
| EBS, basal, BP230 | AR | DST-e /dystonin-e |
| EBS, basal, exophilin 5 | AR | EXPH5 /exophilin |
Generalized Severe Epidermolysis Bullosa Simplex
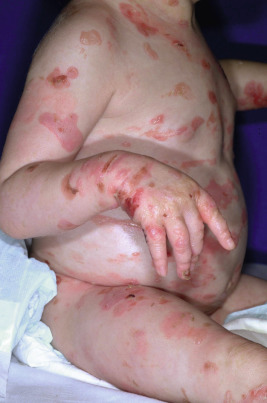
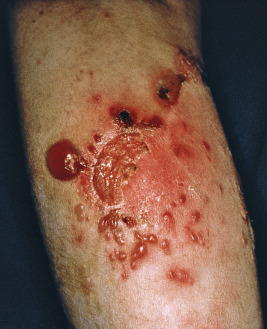
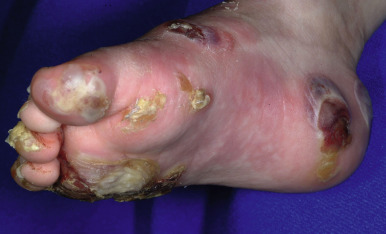
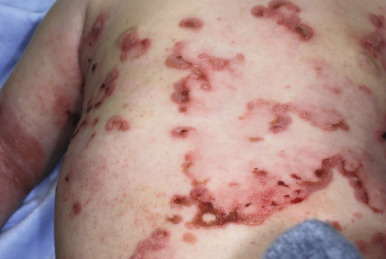
Generalized severe EBS (formerly Dowling–Meara) is the most severe form of EBS from keratin gene mutations ( Table 13-2 ). During the newborn period, the generalized blisters tend to be large and may be difficult to distinguish from those of the severe dystrophic or junctional forms of EB despite the relatively good overall prognosis of babies with this form ( Fig. 13-3 ). Many young infants also show a significant inflammatory reaction in association with blistering and the formation of transient milia at sites of healed blisters, a finding usually characteristic of the dystrophic forms. The characteristic small, clustered (herpetiform) blisters may be seen in neonates, especially on the proximal extremities or trunk, but are more commonly noted during infancy and later childhood ( Fig. 13-4 ). Blistering tends to decrease during later childhood and adulthood, and hyperkeratosis of the palms and soles may develop by 6 or 7 years of age, especially in those younger children with significant palmoplantar blistering ( Fig. 13-5 ). During the neonatal period and early infancy, the extensive blistering may prove life-threatening. After this period, however, the blistering is rarely a threat to life. Nail involvement with sloughing is common in the generalized severe form, but the nails regrow without dystrophy. Similarly, oral and esophageal mucosal blistering may occur and cause problems with feeding and obtaining adequate nutrition, especially with the markedly increased caloric needs of more severely affected infants and younger children. Ocular mucosal blistering is less common, but natal teeth have been described.
| Type | Clinical Manifestations |
|---|---|
| EBS, localized (formerly Weber–Cockayne) | Easy blistering on palms and soles |
| May be focal keratoderma of palms and soles in adults | |
| ≈25% show oral mucosal erosions | |
| Rarely show reticulated pigmentation, especially on arms and trunk and punctate keratoderma (EBS with mottled pigmentation) | |
| EBS, generalized, intermediate (formerly Koebner) | Generalized blistering |
| Variable mucosal involvement | |
| Focal keratoderma of palms and soles | |
| Nail involvement in 20% | |
| Improves with advancing age | |
| EBS, generalized, severe (formerly Dowling–Meara) | Most severe in neonate, infant; improves beyond childhood |
| Large, generalized blisters; later, smaller (herpetiform) blisters | |
| Mucosal blistering, including esophageal | |
| Nails thickened, shed but regrow | |
| May have natal teeth | |
| EBS with mottled pigmentation | Reticulated hyperpigmentation, especially on arms and trunk |
| Punctate keratoses and keratoderma |
Generalized Intermediate Epidermolysis Bullosa Simplex
Generalized intermediate EBS (formerly Koebner EBS) is characterized by generalized blistering of skin, most notable at sites of friction (see Table 13-2 ). Extensive blistering in the neonatal period and early infancy increases the risk of sepsis and may be life-threatening. In general the tendency toward blistering tends to improve with advancing age, particularly by teenage years. Hyperhidrosis is common, and mild to moderate hyperkeratosis of the soles is often present. Although erosions of the mucous membranes may be seen in the newborn as a result of vigorous sucking, mucosal involvement is generally mild and the nails are rarely affected, in contrast to the generalized severe form. Involvement of the conjunctiva and cornea has rarely been described. Migratory circinate EBS results from mutations in the tail domain of KRT5, and is characterized by small blisters on the hands, feet, and legs with migratory circinate erythema ( Fig. 13-6 ) and often postinflammatory hyperpigmentation. There is no mucosal or nail involvement.
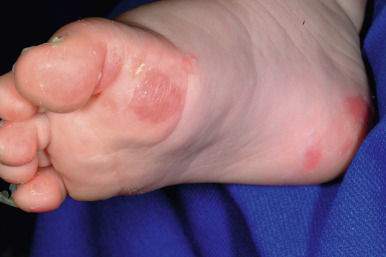
Localized Epidermolysis Bullosa Simplex
Localized EBS (formerly Weber–Cockayne EBS) is the most common clinical variant (see Table 13-2 ). A relatively high threshold of frictional trauma is required to induce blister formation. Bullae are usually confined to the hands and feet (primarily the palms and soles; Fig. 13-7 ); they are often first seen in infants but may not appear until adolescence or early adulthood. The bullae are usually associated with trauma, occur more readily in hot weather with sweating of the feet, and do not tend to be seriously debilitating, although activities that involve trauma to the feet are often restricted. Hyperhidrosis is common, and hyperkeratosis of the palms and soles, although often present, is usually mild. Lesions heal rapidly without scarring, nail involvement rarely occurs, and the mucous membranes do not tend to be involved. In young children, blisters may develop on the knees from the frictional trauma of crawling. In adolescents and young adults, blisters often occur on the feet after long hikes or dancing or on the hands after a game of tennis or golf. EBS with mottled pigmentation is characterized by a mottled, reticulated pigmentation ( Fig. 13-8 ), particularly of the trunk and neck, in association with mild acral blistering (see Table 13-2 ). Patients often show small verrucous papules of the hands and feet and palmoplantar keratoderma.
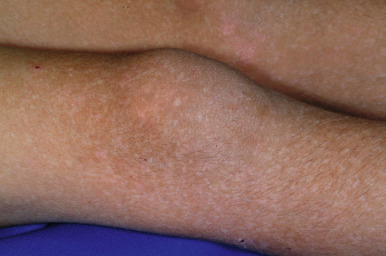
Genetic Basis of Dominant Forms of Basal Epidermolysis Bullosa Simplex
Basal EBS most commonly results from mutations in the genes encoding keratin 5 and keratin 14 (the keratins expressed in basal cells). Keratins are the most abundant proteins of epidermal keratinocytes, and keratin pairs are critical for the filamentous network that provides integrity to epidermal cells. When a point mutation occurs in one of the keratin alleles, resulting in a change in one amino acid, the abnormal keratin protein is still able to form filaments, but these filaments are abnormal. The abnormal filaments do not provide adequate structural integrity to the cell, and as a result, the cell lyses. Cytolysis of epidermal basal cells is the essential histologic feature of all forms of EBS resulting from keratin gene mutations. Electron microscopic examination shows cleavage through the basal layer (above the periodic acid–Schiff-positive basement membrane of the epidermis). In the severe generalized form, clumping of tonofilaments and displacement of nuclei are seen ultrastructurally. The risk of cell lysis and the trauma required to elicit the blistering depend on the site of the mutation and how critical that gene region is for resultant keratin function. The sites of mutations in the most severe generalized form are most critical for keratin function (end-terminal rod domains), whereas the sites mutated in the localized type are least critical. The mutations that lead to EBS with mottled pigmentation tend to be at the head region of KRT5 or KRT14 (most often a mutation that changes the twenty-fifth amino acid of keratin 5), suggesting that this site on the keratin protein is vital for the transfer of pigment from melanocytes to keratinocytes. The migratory circinate form has been linked to mutations in the tail region of KRT5 .
Recessive Forms of Basal Epidermolysis Bullosa Simplex
Basal EBS less commonly is autosomal recessive, including with biallelic mutations in KRT5 or KRT14 (which can phenotypically show localized or generalized blistering). Three clinically distinct forms of EBS result from mutations in the gene-encoding plectin ( PLEC1 ): EBS-Ogna, EBS with muscular dystrophy, and EBS with pyloric atresia. EBS-Ogna, the only disorder with abnormal plectin that is autosomal dominant, mainly shows acral blistering, although more generalized blistering has been described; affected individuals characteristically bruise easily and may show onychogryphosis. EBS with muscular dystrophy presents in the neonatal or infantile period with generalized blisters, but the onset of muscular dystrophy varies from infancy to adulthood. Patients may show ptosis, granulation tissue with stenosis of the respiratory tract, and focal keratoderma. EBS with pyloric atresia presents with the same widespread congenital absence of skin (especially on the extremities), generalized blistering and pyloric atresia as in JEB with pyloric atresia. Malformed pinnae and nasal alae, joint contractures, and cryptorchidism are other shared features. Plectin interacts with α6β4 integrin, providing a rationale for similar clinical manifestations. The mutations of EBS with muscular dystrophy cluster in exon 31, which is spliced out in one of the two isoforms of plectin, explaining the much milder phenotype of EBS with muscular dystrophy. Two mild recessive forms have also been more recently described. EBS with autosomal recessive BP230 deficiency (EBS-AR BP230) results from mutations in encoding dystonin-e (DST-e), the epidermal isoform of bullous pemphigoid (BP) antigen (BPAG1e or BP230). Affected individuals experience localized lifelong blistering and erosions beginning in infancy, but lesions are primarily localized to the ankles and feet. EBS with autosomal recessive exophilin 5 deficiency (EBS-AR exophilin 5) results from mutations in EXPH5, encoding exophilin-5 (or SLAC2-B ), which is a protein involved in vesicle transport. Small, localized erosions and bleeding primary affect site of trauma, primarily the lower back, knees, and ankles. Ultrastructural studies of biopsies provide the clue to diagnosis, showing keratin filament clumping and acantholysis (as can be seen in EBS) but also typical perinuclear cytoplasmic vesicles.
Suprabasal Epidermolysis Bullosa Simplex
To date, biallelic mutations in four genes ( DSP , PKP1 , JUP , and TGM5 ) can lead to autosomal recessive suprabasal forms of EBS. The lethal acantholytic form of EB (LAEB) presents at birth with generalized denudement and absence of hair and nails. Frank blisters are not seen, but the skin peels in sheets ( Fig. 13-9 ). Mucosal sloughing is severe, and affected neonates may be born with teeth. All babies to date have died in the neonatal period. Cardiomyopathy is usually associated. Mutations have been described in the C-terminal domain of DSP . Less deleterious mutations in DSP also cause skin fragility and woolly hair syndrome (see Chapter 7 ), characterized by superficial erosions and crusting with woolly hair, palmoplantar keratoderma, and cardiomyopathy. Acantholytic EBS from mutations in JUP , encoding plakoglobin, manifests at birth as generalized erythroderma with superficial peeling and erosions. Patients have alopecia, dystrophic nails, and recurrent infections and generally die in the neonatal period, but there are no cardiac issues. As with other mutations in desmosomal components, a milder form allows survival but the development of generalized erosions, sparse woolly hair, and focal keratoderma. Plakophilin deficiency (skin fragility and ectodermal dysplasia syndrome) results from mutations in PKP1 , the gene encoding plakophilin-1. Affected patients show generalized erythroderma at birth with blistering. The soles are often most disabling, with palmoplantar keratoderma and painful fissures. Superficial erosions and crusting are prominent in the perioral area, and tongue fissures have been described. The hair tends to be short, sparse and woolly, and the nails are thickened and dystrophic. Affected individuals show variable hypohidrosis, blepharitis, and growth retardation. Plakophilin-1 is a structural component of the desmosomes that allows cell–cell adhesion; biopsies of affected skin show acanthosis, acantholysis, widening of the space between keratinocytes, and few poorly formed desmosomes. Acral peeling skin syndrome, which results from mutations in TGM5 , the gene encoding transglutaminase 5, involves subcorneal cleavage and can be confused with localized EB on the hands and feet. Of note, peeling skin syndrome in an acral or generalized distribution can also result from mutations in other genes (see Chapter 5 ). EBS superficialis involves cleavage of the upper epidermis and stratum corneum in which superficial erosions occur and one can easily induce sheet-like peeling of skin. The mode of transmission and genetic basis remain unclear, but it is classified with the suprabasal forms of EBS.
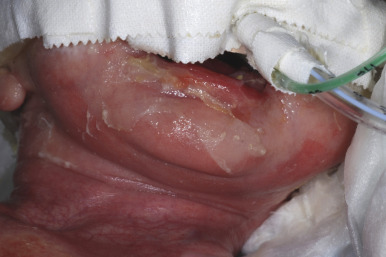
Junctional Epidermolysis Bullosa
Junctional EB (JEB) is group of mechanobullous disorders in which the cleavage plane occurs in the lamina lucida at the junction of the epidermis and dermis ( Table 13-3 ). Encompassing a spectrum from severe life-threatening disease to relatively mild involvement, various subtypes of this disorder have been described, each transmitted in an autosomal recessive manner ( Table 13-4 ). Autosomal dominant disorders (mutation on one allele) with only dental manifestations from a missense mutation in LAMB3, dental issues and blistering from a missense mutation in COL17A1, or the corneal erosions from a missense mutation in COL17A1 (termed epithelial recurrent erosion dystrophy [ ERED ]) have been described.
| Type | Inheritance | Gene Defect |
|---|---|---|
| JEB, generalized severe (formerly Herlitz) | AR | LAMA3, LAMB3, LAMC2/ laminin 332 |
| JEB, generalized, intermediate (formerly non-Herlitz) | AR | Mild mutation: laminin 332 |
| JEB, generalized, intermediate (formerly non-Herlitz) | AR | COL17A1 /type XVII collagen |
| JEB, generalized with pyloric atresia | AR | ITGA6, ITGB4 /integrin α6 or β4 |
| JEB, generalized, late onset | AR | COL17A1 /type XVII collagen |
| JEB, generalized, with respiratory and renal involvement | AR | ITGA3/ integrin α3 |
| JEB, localized | AR | COL17A1 /type XVII collagen |
| ITGA6, ITGB4 /integrin α6β4 | ||
| laminin 332 | ||
| JEB with pyloric atresia | AR | ITGA6, ITGB4 /integrin α6 or β4 |
| JEB, localized, inversa | AR | COL17A1 /type XVII collagen |
| LOC syndrome | AR | Laminin 332, α3 chain |
| Type | Clinical Manifestations |
|---|---|
| JEB, generalized severe (formerly Herlitz) | 50% of patients die by 2 years old |
| Blisters heal with atrophic scarring but no milia | |
| Periungual and fingerpad blistering, erythema | |
| Blistering of oral and esophageal mucosae | |
| Laryngeal and airway involvement with early hoarseness | |
| Later, perioral granulation tissue with sparing of lips | |
| Anonychia | |
| Dental enamel hypoplasia, excessive caries | |
| Growth retardation | |
| Anemia | |
| JEB, generalized, intermediate (formerly non-Herlitz) | Less severe, but similar manifestations to Herlitz type, including dental, nail and laryngeal involvement |
| Granulation tissue is rare | |
| Perinasal cicatrization | |
| Less mucosal involvement | |
| Alopecia | |
| Anemia but not as severe as JEB, generalized severe | |
| JEB, localized | Localized blisters without residual scarring or granulation tissue |
| Minimal mucosal involvement | |
| Dental and nail abnormalities as in JEB, generalized severe | |
| JEB, generalized with pyloric atresia | Usually lethal in neonatal period |
| Generalized blistering, leading to atrophic scarring | |
| May be born with large areas of cutis aplasia | |
| No granulation tissue | |
| Nail dystrophy or anonychia | |
| Pyloric atresia, genitourinary malformations | |
| Rudimentary ears | |
| Dental enamel hypoplasia (survivors) | |
| Variable anemia, growth retardation, mucosal blistering |
Early diagnosis of the subtype of JEB is critical and is based on immunomapping (including detection of level of cleavage and lamina lucida proteins) and if available, genotyping. Immunomapping studies of sections from skin adjacent to a freshly induced blister show cleavage at the lamina lucida level. Electron microscopic evaluations reveal markedly reduced or absent hemidesmosomes, anchoring structures that span the lamina lucida of the basement membrane of skin and mucosae.
In the severe generalized form of JEB (formerly called JEB-Herlitz ), blistering begins at birth and death occurs in 45% by 1 year of age and 54% by 2 years of age. Survival into adulthood is common for most other forms of JEB but has been occasionally described for individuals with the severe generalized type of JEB.
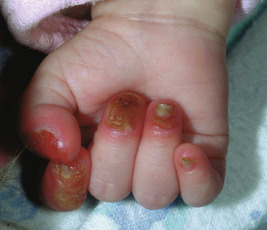
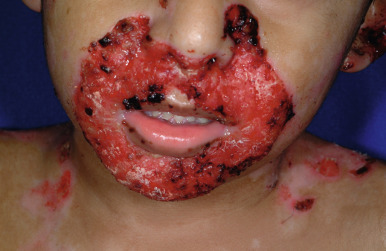
Severe generalized JEB is characterized by the generalized distribution of blisters and large erosions mainly on the buttocks, perioral area, trunk, and scalp. Blistering is almost always present at birth (see Fig. 13-2 ). Blistering of the fingertips with sloughing of the nails ( Fig. 13-10 ) and perioral involvement ( Fig. 13-11 ) with sparing of the lips are important if not diagnostic features of the junctional forms of EB. Granulation tissue, especially of the perioral region, is characteristic of this severe form (and laryngoonychocutaneous [LOC] syndrome), and is usually present by a few years of age. Sites of healing tend to be atrophic but show far fewer milia than the dystrophic forms. The mucous membranes are affected, especially the oral mucosae. Hoarseness and laryngeal involvement are common, and airway involvement may lead to death. The genitourinary and gastrointestinal tracts are often affected as well. The teeth are dysplastic, and a cobblestone appearance to the dental enamel is characteristic. Severe growth retardation and recalcitrant anemia are common. Mutations in this severe generalized form always affect a gene encoding one of the three chains of laminin 332 ( LAMA3, LAMB3, and LAMC2 encoding α3, β3, and γ2 chains, respectively).
Generalized intermediate or localized JEB, may also result from less deleterious mutations in one of the laminin 332 genes. Mutations in the α3 chain of laminin 332 can lead to the localized form, LOC syndrome (Shabbir syndrome). LOC syndrome occurs most often in the Punjab region of India and Pakistan. It features excessive granulation tissue of the larynx (leading to hoarseness and potentially airway obstruction), conjunctival granulation tissue (leading to symblepharon, corneal scarring, and potentially blindness), and erosive blisters most commonly on the face and neck. Death is common because of respiratory-tract involvement. JEB inversa presents with blisters predominantly in intertriginous areas (axillary areas and groin). Mucosal blistering tends to be variable in extent but less than in the generalized forms. A rare form, JEB with respiratory and renal involvement, shows limited skin blistering on the legs and buttocks but congenital nephrotic syndrome and severe neonatal respiratory distress with interstitial pneumopathy. The disorder results from biallelic mutations in the gene encoding integrin α3, and affected infants die within the first months of life.
Generalized intermediate JEB (previously called non-Herlitz JEB or generalized atrophic benign EB [ GABEB ]) most commonly affects the gene encoding BP180 (collagen XVII; COL17A1 ) but occasionally reflects at least one missense mutation in a gene encoding laminin 332. In general, the features tend to be less severe than in the severe generalized form, but granulation tissue is rarely seen and EB nevi are common. EB nevi are dark, irregular hyperpigmented patches that may be worrisome dermoscopically but show benign nevi or increased basal pigment deposition histologically and clear spontaneously during the subsequent months to years. Diffuse alopecia (scarring or nonscarring) is a key feature of this form, and narrowing of the nares is often seen. Squamous cell carcinoma (SCC) occasionally occurs in affected areas later in life; the occurrence of these features that are not typically seen in the severe generalized form may reflect the greater lifespan in this form.
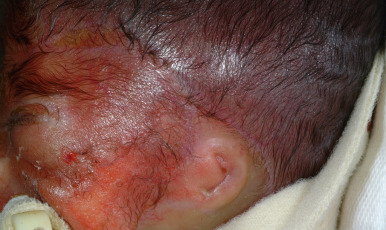
JEB with pyloric atresia results from mutations in either α6 integrin ( ITGA6 ) or its hemidesmosome partner, β4 integrin ( ITGB4 ). Blistering is generalized at birth, often with large areas of cutis aplasia. Other features in addition to pyloric atresia are rudimentary and malformed ears ( Fig. 13-12 ) and genitourinary tract malformations, attesting to the important role of α6β4 integrin in the development of the ears, pylorus, and genitourinary tract. Less commonly, EB with pyloric atresia results from plectin abnormalities. A late-onset form of JEB has been described with onset in young adulthood or later; nails are dystrophic or absent and dental enamel is hypoplastic, but blistering is milder than in other junctional forms. Patients may have hyperhidrosis and absent dermatoglyphics.
Dystrophic Epidermolysis Bullosa
The scarring (dystrophic) types of EB are predominantly divided into dominant dystrophic epidermolysis bullosa (DDEB) and recessive dystrophic epidermolysis bullosa (RDEB) forms with varying degrees of severity ( Tables 13-5 and 13-6 ). The recessive forms have been further subdivided into the severe generalized and generalized other forms. In general, the dominant forms are considerably less severe; affected individuals are generally healthy, are of normal stature, and show limited blistering of the skin. The severe generalized form of RDEB, conversely, is severe and incapacitating. Functional deformities of the hands and feet result from extensive scarring, growth and development are retarded, and profound anemia and hypoalbuminemia are standard. All forms of DEB affect anchoring fibrils, critical elements for epidermal–dermal cohesion, and result from mutations in type VII collagen.
| Type | Inheritance | Gene Defect |
|---|---|---|
| Dominant | ||
| DDEB, generalized | AD | COL7A1 /collagen VII |
| DDEB, rare types: acral, pretibial, pruriginosa, nails only, bullous dermolysis of newborn | AD | COL7A1 /collagen VII |
| RDEB, generalized severe | AR | COL7A1 /collagen VII |
| RDEB, generalized intermediate | AR | COL7A1 /collagen VII |
| RDEB, inversa | AR | COL7A1 /collagen VII |
| RDEB, rare types: localized, pretibial, pruriginosa, centripetalis, bullous dermolysis of the newborn | AR | COL7A1 /collagen VII |
| Type | Clinical Manifestations |
|---|---|
| Dominant dystrophic | Onset at birth to early infancy |
| Blistering predominates on dorsum of hands, elbows, knees, and lower legs | |
| Milia associated with scarring | |
| Some patients develop scar-like lesions, especially on the trunk | |
| 80% have nail dystrophy | |
| Recessive dystrophic, severe generalized | Present at birth |
| Widespread blistering, scarring, milia | |
| Deformities: pseudosyndactyly, joint contractures | |
| Severe involvement of mucous membranes, nails; alopecia | |
| Growth retardation, poor nutrition | |
| Anemia | |
| Mottled, carious teeth | |
| Osteoporosis, delayed puberty, cardiomyopathy, glomerulonephritis, renal amyloidosis, IgA nephropathy | |
| Predisposition to squamous cell carcinoma in heavily scarred areas | |
| Recessive dystrophic, generalized intermediate | Generalized blisters from birth with milia, scarring |
| Less anemia, growth retardation, mucosal but more esophageal issues with advancing age |
Dominant Dystrophic Epidermolysis Bullosa
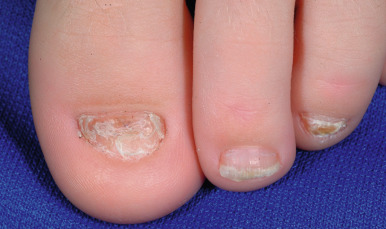
Generalized dominant dystrophic epidermolysis bullosa (DDEB) usually presents at birth or shortly thereafter, although mild cases may not show blistering or nail changes until adulthood. The blisters and resultant scars and milia formation primarily involve the extensor areas of the extremities and the dorsum of the hands (see Fig. 13-15 ). Nail thickening, dystrophy, or complete nail destruction are seen in 80% of cases ( Fig. 13-13 ). Although mucous membrane lesions appear in 20% of cases, they tend to be mild and not problematic. The teeth and hair are generally not affected, and physical development is normal. Some forms of DDEB are localized, including localized to the nails (DDEB, localized, nails only) or acral areas and/or hands and feet. The pretibial form of DDEB shows lichenoid or atrophic papules and plaques on the pretibial areas but also hand, foot, and nail involvement. DEB pruriginosa (DEB-Pr) is a poorly understood form of DEB (usually dominant) with the onset of severe associated pruritus and prurigo-like, scarred vesicles and papules primarily of the pretibial areas that is often delayed until adolescence or adulthood. Bullous dermolysis of the newborn (DEB-BDN; formerly called transient BDN ) shows skin blistering, often extensive, at birth or in early infancy ( Fig. 13-14 ). However, blistering dramatically improves during the first months to 2 years of life, and beyond mild residual atrophy, scarring, and nail dystrophy and an increased risk of dental caries, ongoing blistering is not a problem. The disorder results from mild mutations in COL7A1 and can be inherited in a dominant or recessive manner.
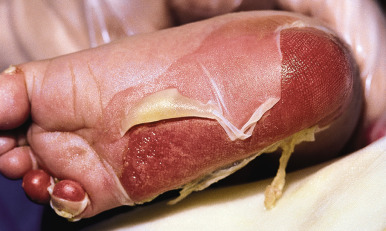
Severe Generalized Recessive Dystrophic Epidermolysis Bullosa
Children with severe generalized RDEB (formerly called the Hallopeau–Siemens type ) have a severe life-altering bullous disease characterized by widespread dystrophic scarring and deformity and by severe involvement of mucous membranes. RDEB may manifest as a less severe form (generalized intermediate RDEB) with less severe blistering of the skin and mucosae that may be mistaken for DDEB ( Fig. 13-15 ); individuals with this form often begin to have problems with esophageal function in adolescence. Two other forms of RDEB have more localized cutaneous involvement. Blistering in RDEB inversa tends to involve the intertriginous axial, lumbosacral, and acral sites in addition to extensive mucosal involvement, including esophageal strictures and external auditory canal stenosis. RDEB centripetalis is a rare RDEB form that starts with limited involvement (hands, feet, nail dystrophy) but progresses with an advancing border to involve most of the extremities by mid-adulthood; face, truncal, and mucosal involvement never occur.
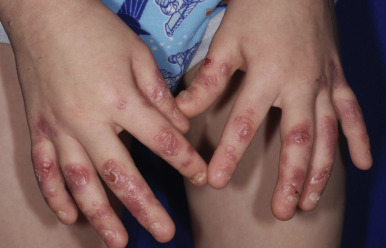
Although any area of the skin may be involved in infants with generalized severe RDEB, the most commonly affected areas are the hands, feet, buttocks, scapulae, face, occiput, elbows, and knees. In older children the hands, feet, knees, elbows, and posterior neck and/or upper mid-back ( Fig. 13-16 ) are most commonly involved. Bullae may be hemorrhagic, and large areas, especially on the lower extremities, may be completely devoid of skin. When a blister ruptures or its roof peels off, a raw painful surface is evident. The Nikolsky sign (production or enlargement of a blister by slight pressure or the production of a moist abrasion by slight pressure on the skin) is often positive. Fluid contained in bullae, although at first sterile, may become secondarily infected, which can lead to sepsis; Staphylococcus aureus and Pseudomonas aeruginosa are the most common organisms.
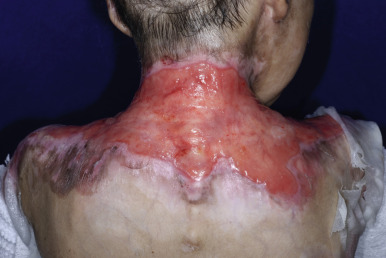
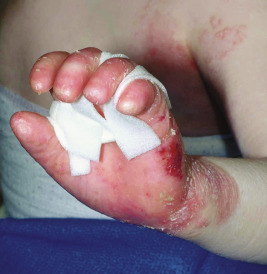
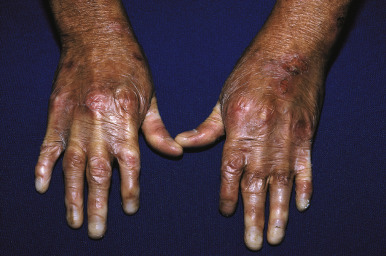
Bullae are often followed by atrophic scars and varying degrees of hyperpigmentation or hypopigmentation. Milia overlying the scars are characteristic. EB nevi are common (see above). The hands and lower aspects of the legs are particularly susceptible to severe blistering and scarring. The fingers and toes may become fused, with resultant pseudosyndactyly in which the digits become bound together by a glove-like epidermal sac, with resulting claw-like clubbing or mitten-like deformities ( Fig. 13-17 ). The fingers and toes become immobile (usually during the first years of life), and the wrists, elbows, knees, and ankles may become fixed in a flexed position from contractures, leading to immobility and often confinement to a wheelchair.
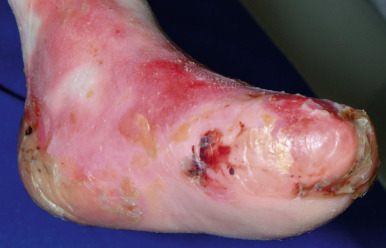
Oral mucosal involvement occurs soon after birth ( Fig. 13-18 ), leading to dysphagia and limiting the ability to suck well. Erosions of the esophagus may at times result in segmental stenosis (most often in the upper third) with consequent difficulty in swallowing. Gastroesophageal reflux disease often occurs, especially presenting as effortless vomiting. Constipation is common and may be related to anal fissuring, inadequate dietary fiber, and administration of iron. Affected children are reluctant to eat and often fail to thrive, given their increased nutritional needs owing to loss of protein and other nutrients through wounds. As the child grows older there is a tendency for the disease to become less severe, but the affected individual soon learns to avoid hot drinks, rough foods, and large particles that might produce blistering of the mouth, pharynx, or esophagus. Typically, patients show microstomia owing to intraoral scarring and a frenulum that is bound down. The eyes may develop blisters with associated ocular inflammation and later corneal scarring, potentially leading to visual impairment. Hoarseness, aphonia, and even laryngeal stenosis may result from laryngeal blistering and scarring. Bone mineralization is low in patients with EB, particularly those with RDEB, probably owing to a combination of insufficient nutrition, reduced physical activity, and chronic inflammation.
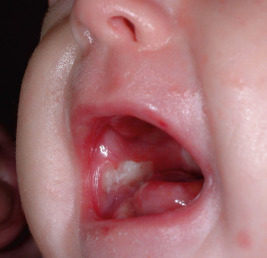
The teeth in RDEB are particularly susceptible to early and commonly severe caries. The progressive intraoral scarring leads to microstomia and decreased salivation. Even routine dental care may cause the eruption of bullae and erosions on the lips, gingivae, and oral mucosa. The nails may show severe dystrophy or complete absence of nails. Scalp and body hair may be sparse, and there may be patches of cicatricial alopecia.
In patients with the severe generalized form of RDEB, death may occur during infancy or childhood as a result of septicemia, pneumonia, or renal failure. Patients with RDEB (and rarely DDEB) have an increased risk of glomerulonephritis, renal amyloidosis, and immunoglobulin (Ig) A nephropathy. The tremendous loss of fluid, blood, and protein through the skin coupled with malnutrition can lead to hypoalbuminemia and anemia. Dilated cardiomyopathy is an uncommon complication (4.5% by 20 years of age) but may be fatal, especially in the presence of concurrent chronic renal failure. The cause of the cardiomyopathy may be multifactorial, including from transfusion-associated iron overload, viral myocarditis, and deficiency of selenium and carnitine. Other complications of RDEB include erosions and scarring of the anal area (often resulting in severe discomfort, chronic constipation, or soiling), urethral stenosis, urinary retention, hypertrophy of the bladder, and occasionally hydronephrosis.
Patients with RDEB (and to a lesser extent JEB but not DDEB) show a progressively increasing risk of developing cutaneous SCCs (7.5%, 68%, 80%, and 90% by 20, 35, 45, and 55 years of age, respectively) in heavily ulcerated and scarred areas of skin. These lesions are predominantly over joints and on the distal extremities and present as nodular lesions or nonhealing ulcers. Suspicious masses should be biopsied to distinguished SCCs from benign lesions such as verruciform xanthoma ; MMP13 expression is strongly positive in SCC but is negative in benign hyperkeratotic lesions. Cutaneous carcinomas tend to be locally aggressive, often requiring amputation, and tend to metastasize, leading to death.
Death during childhood is most common with JEB (median age 4 to 6 months). Sepsis, failure to thrive, and respiratory failure are the major causes of death during childhood. Children with RDEB generally survive the neonatal and infantile periods but succumb to infection later during childhood or to aggressive cutaneous carcinomas during adulthood.
Kindler Syndrome
Kindler syndrome is characterized by generalized progressive poikiloderma, congenital acral skin blistering, diffuse cutaneous atrophy ( Fig. 13-19 ), skin fragility, webbing of the fingers and toes with digital tapering, nail dystrophy, oral mucosal lesions, and photosensitivity, sometimes within minutes after exposure. Other features are hyperkeratosis of the palms and soles; leukokeratosis; ectropion; red friable hyperplastic gums; constipation and sometimes severe colitis; esophageal, laryngeal, anal, vaginal and urethral meatal stenosis; and phimosis. Although the photosensitivity and the blister formation seem to decrease with age, the atrophic scarring and poikiloderma increase. The incidence of SCC of the acral skin or mouth is increased after the age of 30 years. Because the blistering arises at multiple levels within and/or beneath the basement membrane zone, Kindler syndrome has been classified as mixed EB. Treatment of this disorder requires the avoidance of trauma and the proper use of emollients, appropriate sun protection, and the judicious use of antibiotics to prevent secondary infection. Regular dental care and surveillance for early malignancies are important, as are iron replacement if the patient is anemic and management of the stenoses and colitis. The gene mutated in Kindler syndrome is FERMT1 (formerly called KIND-1 ), which encodes fermitin family homolog 1 (FFH1) protein or kindlin-1, a focal adhesion protein that regulates keratinocyte cell adhesion, motility, and stem cell homeostasis.
Treatment of Epidermolysis Bullosa
As in any inherited disorder, it is the responsibility of the physician to inform parents of the risks of transmitting genetic abnormalities. When the condition is determined by a dominant gene (as in DDEB) and a parent is affected, the risk of the disorder in siblings is 50%. In a family in which a child manifests abnormalities because of a recessive gene (as in RDEB), parents risk a 25% possibility with each pregnancy of the disorder occurring in future offspring. Appropriate genetic counseling, however, depends on accurate diagnosis. Since the clinical course of many forms of EB is variable, especially during the neonatal and infantile periods, it is recommended that patients be carefully evaluated as early as possible with immunofluorescent mapping, monoclonal antibody studies, and deoxyribonucleic acid (DNA) analysis if appropriate in an effort to establish the correct diagnosis. Prenatal diagnosis of all forms of EB is now available using molecular techniques, but is easiest if the gene defect is known in that family. The current availability of whole exome sequencing has allowed concurrent investigation of several types of EB for a fraction of the price of sequencing any one individual gene. Preimplantation diagnosis has been performed and is an option that utilizes in vitro fertilization to ensure a normal fetus without the risk of abortion.
The psychosocial effects of EB, especially the more severe forms, on the affected individual and family are among the most dramatic of any skin disease. Affected children are concerned about having itchy skin, being in pain, having difficulty with participation, failing to understand others, and feeling different. Parents of affected children worry about the child being different, the child suffering pain, feeling uncertain about the future, restrictions on employment and leisure, problems with organizing care, being constantly on duty, family problems, the ignorance and lack of skills of alternative care providers, and resistance by the child to care. These problems should be discussed and psychological support for patients and their families offered as part of optimal care.
The treatment of EB is largely palliative, with protection from friction or overheating, avoidance of abrasion and constriction, control of secondary infection, nutritional supplementation, and pain control. Because blisters result from mechanical injury, measures should be taken to relieve pressure and prevent unnecessary trauma. Clothing should be soft and worn inside out. Labels that may rub the skin should be removed. Velcro closures are less traumatizing than other traditional closures. Mittens can be worn to minimize self-induced trauma. Shoes should be soft and fit well; leather shoes with leather linings, ideally with external seams (e.g., moccasins), are usually recommended. During the summer, canvas shoes and jelly sandals are the best choices. Shoes should be large enough to accommodate dressings and minimize friction. Insoles can be made from cooling gel, sheepskin, or protective dressings. Affected babies can be lifted and moved on a soft pad, and the bathtub can be lined with a thick towel. A cool environment and lubrication of the skin to decrease surface friction are helpful in the reduction of blister formation. When blisters occur, extension may be prevented by aseptic aspiration of blister fluid. The roofs of blisters should be left intact whenever possible to protect the underlying skin.
Keeping palms and soles with EBS cool and dry helps to minimize blistering, especially during hot weather. Hyperhidrosis is often a concomitant feature, and measures to minimize the increased blistering associated with hyperhidrosis can be helpful. These include applying 20% aluminum chloride hexahydrate at night and gently drying the area with a cool hair dryer, wearing socks that absorb moisture, and sprinkling of affected areas with absorbent powder such as Zeasorb. For extreme cases and in older patients with the localized form of EBS, injections of botulinum toxin A have been advocated. Silver-impregnated socks can decrease infections and increase foot comfort.
A water mattress and a soft fleece covering will help to limit friction and trauma. Daily baths and topical application of protective petrolatum to eroded areas or, especially if slightly crusted, antibiotic ointments (usually bacitracin, mupirocin, or gentamicin) are helpful. Protective dressings that do not adhere to wounds should be applied to eroded areas to promote healing but prevent further denudation when dressings are changed (e.g., petrolatum-impregnated gauze, Telfa, Mepilex, Mepilex Transfer, Mepitel, Restore). In children with RDEB, dressings should be carefully placed between the digits, although the ameliorative effect of this practice to decrease the risk of pseudosyndactyly ( Fig. 13-20 ) has not been tested. Sterile precautions must be taken when changing dressings to reduce the risk of bacterial infection. Tape and any significant pressure to skin must be avoided. Dressings can be held in place by rolled gauze (such as Kerlix) or loose self-adherent wraps) with tape only applied to the dressing itself or by stockinette (such as Surgifix or Spandage). Dressings with silver have helped patients with recurrent infections, but application of silver sulfadiazine has been associated with argyria. Families need to weigh the antiinfective benefits with the risk of potentially high blood levels of silver. Bacteriostatic dressings with methylene blue and crystal-violet dyes (such as Hydrofera blue) and medical-grade honey are also available. Crusted or purulent areas should be cultured and treated based on the sensitivity of organisms. Topical application of mupirocin and/or gentamicin ointments may be useful for limited areas of crusting (see mention of readthrough with gentamicin ointment below). More extensive involvement requires administration of systemic antibiotics. Excessive usage of systemic antibiotics, however, should be avoided because of the high risk of development of resistance. Gentamicin soaks (480 mg/L saline), acetic-acid soaks (diluted white vinegar), and addition of small amounts of bleach to the bathwater (e.g., ![]() to
to ![]() cup per tub) have been used to decrease the overgrowth of Pseudomonas and staphylococcal organisms. The risk of sepsis with cutaneous infection is high in neonates and infants, and patients should be monitored carefully. Topical and systemic steroids are generally not useful for patients with EB and should be avoided in view of their promotion of infection and other side effects. However, limited application of potent topical steroid, or oral thalidomide has been helpful for the granulation tissue of laminin 332 defects.
cup per tub) have been used to decrease the overgrowth of Pseudomonas and staphylococcal organisms. The risk of sepsis with cutaneous infection is high in neonates and infants, and patients should be monitored carefully. Topical and systemic steroids are generally not useful for patients with EB and should be avoided in view of their promotion of infection and other side effects. However, limited application of potent topical steroid, or oral thalidomide has been helpful for the granulation tissue of laminin 332 defects.