Abstract
Immunobullous diseases, while rare, may present severe medical and therapeutic challenges to the treating clinician. These entities include pemphigus and its variants, bullous pemphigoid and its variants, epidermolysis bullosa acquisita, linear IgA bullous dermatosis, and dermatitis herpetiformis. Classification and treatment require an understanding of the basic biology of keratinocyte to keratinocyte and keratinocyte to extracellular matrix interactions as well as the disease-specific autoantibodies that interfere with these relationships. Standard histology in combination with direct and indirect immunofluorescence testing provides the cornerstone for diagnosis. Significant strides have been made within the last decade in the development of non-steroidal medications with the introduction of IVIg and biologic therapies such as rituximab. Improved therapeutics have resulted in decreased mortality and morbidity from the disease states themselves as well as from treatment-induced complications.
Key words
Azathioprine, Bullous pemphigoid, Dapsone, Dermatitis herpetiformis, Epidermolysis bullosa acquisita, IVIg, Linear IgA bullous dermatosis, Mycophenolate mofetil, Pemphigus, Rituximab
- •
Immunobullous diseases are a complicated multisystem management challenge requiring advanced immunosuppressive regimens.
- •
Pathogenesis has been linked to disease-specific autoantibodies to structural antigens within the epidermis and basement membrane zone.
- •
The location of blistering, risk of subsequent scarring, and degree of systemic involvement depend upon the type and distribution of antigen targeted.
- •
Clinical presentation, histologic findings, and immunofluorescence studies (both indirect and direct) are the cornerstone of diagnosis.
- •
Treatment of pemphigus and pemphigoid includes systemic steroids, azathioprine, mycophenolate mofetil, and rituximab, as well as IVIg or plasmapheresis for refractory cases.
- •
Epidermolysis bullosa acquisita is notoriously refractory to treatment, but may respond to management similar to that of bullous pemphigoid.
- •
Drug-induced and paraneoplastic variants of pemphigoid, pemphigus, and linear IgA bullous dermatosis occur that tend to improve following cessation of the offending drug or treatment of the underlying malignancy.
- •
Dermatitis herpetiformis is a manifestation of celiac disease and responds to a strict gluten-free diet.
- •
Because dermatitis herpetiformis and linear IgA bullous dermatosis are both neutrophil-mediated processes, dapsone is the most effective immunomodulating agent.
Immunobullous diseases can precipitate extreme stress to multiple organ systems. Severe cases can have extensive mucocutaneous involvement, and have the potential for secondary infection and fluid loss. These diseases are also associated with a variety of systemic disorders, both directly and indirectly. Furthermore, the requirement for systemic corticosteroid and immunosuppressive therapy may produce myriad systemic complications.
Comprehension of the biology of keratinocyte interactions with other keratinocytes and with the extracellular matrix is essential for understanding the autoimmune blistering diseases. Keratinocytes attach to each other by desmosomes, whereas they attach to the underlying basement membrane by hemidesmosomes ( Fig. 16-1 ). Desmosomes consist of an intracellular cytoplasmic plaque made up of the molecules desmoplakin, plakophilin, and plakoglobin. This cytoplasmic plaque interacts with the intracellular scaffolding of tonofilaments and anchors the keratin intermediate filaments to the hemidesmosome. The extracellular portion of the desmosome is comprised of desmogleins and desmocollins, which project onto the cell surface of the keratinocyte and contact desmosomal proteins of neighboring keratinocytes. Disruption of desmosomal interactions leads to acantholysis.
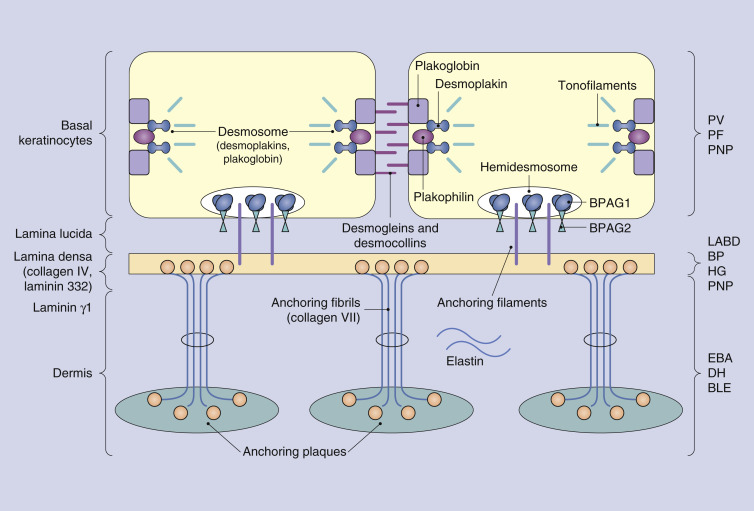
Hemidesmosomes attach basal keratinocytes to the underlying basement membrane. Together with the basement membrane zone (BMZ), hemidesmosomes form the dermoepidermal junction. Proteins that comprise the hemidesmosome are the bullous pemphigoid antigens 1 (BP230) and 2 (BP180), plectin, and α6β4 integrin. BPAG1 is a 230-kDa intracellular molecule, whereas the 180-kDa BPAG2 is an anchoring filament that spans the keratinocyte extracellular membrane, extending into the BMZ. Disruption of hemidesmosomal interactions is implicated in bullous pemphigoid, pemphigoid (herpes) gestationis (discussed in Chapter 35 ), and linear IgA bullous dermatosis. The BMZ is divided into the lamina lucida and lamina densa, named according to their transmission electron microscopic appearance. Anchoring filaments extend from the hemidesmosome through the lamina lucida to the lamina densa, where they interact with collagen IV and laminin 332. The papillary dermis attaches to the BMZ by anchoring fibrils, which either extend from the lamina densa down to dermal anchoring plaques, or loop back up to reattach to the lamina densa. Anchoring fibrils are composed of collagen VII. The blistering in the epidermolysis bullosa acquisita is thought to be due to disruption of molecular interactions in this region. Important structural proteins in the dermis include types I and II collagen and elast in.
Pemphigus
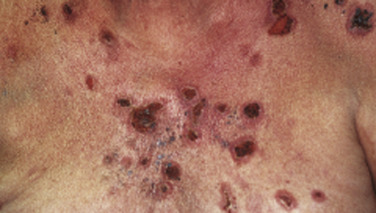
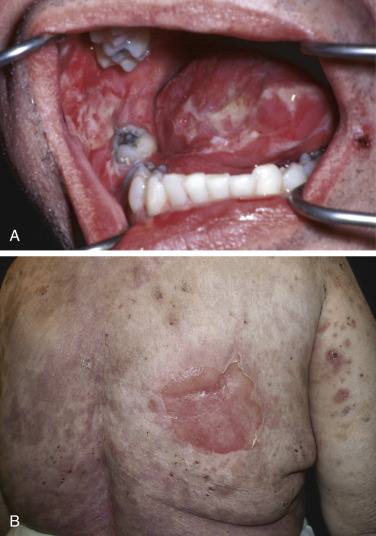
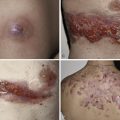
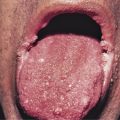
Pemphigus is characterized by detachment of adhesions between keratinocytes (acantholysis). This process results in vesicles, bullae, and subsequent erosions of both cutaneous and mucosal surfaces. Affected skin in pemphigus vulgaris reveals flaccid blisters that generally develop on noninflamed skin, are readily broken, and progress to large, weeping, denuded areas ( Fig. 16-2 ). Oropharyngeal erosions are common and may be the presenting sign ( Fig. 16-3 ). The pemphigus group of diseases is divided into pemphigus vulgaris (with its variant pemphigus vegetans), pemphigus foliaceus (with its variants pemphigus erythematosus and fogo selvagem), IgA pemphigus, and paraneoplastic pemphigus.
Pathogenesis
The pathogenetic process in pemphigus is that of an organ-specific autoimmune disease. Lesional skin and usually serum demonstrate the presence of an IgG class autoantibody directed against desmoglein antigens present in normal squamous epithelium. The exact mechanism for this loss of self-tolerance is unknown, but CD4+ T cells recognize distinct epitopes of the extracellular portions of desmoglein 1 and 3, and preferentially produce T-helper type 2 (Th2) cytokines. Autoantibody production in pemphigus vulgaris and pemphigus foliaceus is polyclonal, and most antibodies in active disease are of the IgG 4 subclass. Patients in remission who have persistent pemphigus antibodies in their serum have mainly the IgG 1 subtype.
Several lines of evidence support the critical role of antibodies to desmogleins in the clinical acantholytic process. First, the antibody is consistently present in lesional skin and the patient’s serum. The serum autoantibody titers correlate with disease activity and there is a therapeutic response to plasmapheresis. Passive transfer of pemphigus from mothers to neonates occurs. Pemphigus antibody produces acantholysis when added to normal human skin in organ culture, and results in detachment when added to epidermal cell cultures. Further convincing evidence of the pathogenetic role of pemphigus antibody has been provided by the demonstration that IgG fraction purified from the serum of patients with pemphigus can induce a disease in neonatal mice that reproduces the clinical, histologic, and immunologic features of human pemphigus.
There are two alternative hypotheses to explain the autoantibody-mediated acantholysis: the desmoglein compensation hypothesis and the intracellular signaling hypothesis. In the desmoglein compensation hypothesis, binding of IgG to desmoglein molecules structurally disrupts epidermal cell adhesion. Desmoglein 1 (Dsg 1) expression is very low in mucosa, but it is expressed throughout the skin and increases in the more superficial layers. Desmoglein 3 (Dsg 3), by contrast, is expressed in all levels of mucosa, but only in and near the basal layer of the skin. Therefore, pemphigus foliaceus with Dsg 1 antibodies leads to superficial blistering in the skin, but no oral involvement. Pemphigus vulgaris with only Dsg 3 antibodies (mucosal-dominant pemphigus vulgaris) leads to oral erosions but minimal skin blisters. Amagai has proposed the “desmoglein compensation theory” as an explanation of this phenomenon, suggesting that Dsg 1 in the skin can “compensate” for the loss of Dsg 3 adhesion in this form of the disease. Finally, there can be no desmoglein compensation in pemphigus with both Dsg 1 and 3 antibodies (mucocutaneous pemphigus vulgaris), so acantholysis results in both skin and mucosa. Complement activation is not thought to play a role in acantholysis. The second hypothesis involves binding of IgG to the desmoglein cell surface antigen. This triggers transmembrane signaling and a series of intracellular pathways that lead to separation of the epidermal cells. These pathways are complex, but the validity of the process has been documented in vitro.
Although the stimulus for pemphigus antibody formation is unknown, the endemic nature of pemphigus foliaceus in Brazil suggests the involvement of an environmental factor, possibly an infectious agent. Most of the cases in Brazil occur in populations residing near rivers, and an insect vector for a microorganism has been proposed. Whether pemphigus in other geographic areas is precipitated by similar events is unknown. d -Penicillamine may produce pemphigus (predominantly pemphigus foliaceus) in patients being treated for rheumatoid arthritis, Wilson’s disease, scleroderma, or other penicillamine-responsive disorders. Consequently, it is likely that a variety of stimuli may give rise to epidermal antigen intolerance.
There is a strong association of pemphigus vulgaris with the HLA class II alleles HLA DRB ∗ 0402, DRB ∗ 0401, and DQB1 ∗ 0503. These HLA alleles may restrict autoreactive responses to desmoglein 3.
Classification
There are two histopathologically distinct forms of pemphigus. Pemphigus vulgaris is the most severe type and is characterized histopathologically by suprabasilar cleft formation, whereas pemphigus foliaceus is less severe and is distinguished by blister formation within or just beneath the granular layer. In pemphigus vulgaris and, to a lesser extent, pemphigus foliaceus, the blister may extend into surrounding nonblistered skin by applying shearing pressure to perilesional tissue (Nikolsky’s sign).
Pemphigus vulgaris is the most common form of pemphigus and is generally seen in the fourth to sixth decades of life. It has, however, been described both in children and in the elderly. Prior to the use of corticosteroids, 50% of patients with the disease died within the first 12 months, most frequently from secondary cachexia, sepsis, and/or electrolyte imbalance. Now, with the broad utilization of immunosuppressants, the mortality is 5%.
Pemphigus vegetans is a rare variant of pemphigus vulgaris. Proliferative and verrucous lesions with surrounding pustules gradually develop from denuded bullae on intertriginous cutaneous surfaces. Such lesions were more common in the era before corticosteroid use and may represent a host response to the blistering process. Spontaneous remission is somewhat more likely in the vegetans form. Histopathologically one sees epidermal proliferation with hyperkeratosis, papillomatosis, and acantholysis with intraepidermal abscesses containing eosinophils.
Pemphigus foliaceus is generally less severe than pemphigus vulgaris. The superficial vesicles rupture easily, producing shallow erosions and crusting that clinically resemble impetigo. Clinical blistering may be totally absent. Lesions occur on the chest, back, and scalp, and may produce a seborrhea-like scaling with eventual spread to acral areas after a prolonged period. Clinically visible mucosal lesions are absent, even in advanced cases. Autoantibodies in pemphigus foliaceus are directed predominantly against desmoglein 1. Pemphigus herpetiformis is a morphologic variant of pemphigus vulgaris or pemphigus foliaceus that occurs as grouped vesicles.
Fogo selvagem is an endemic form of pemphigus foliaceus seen in Brazil. It occurs predominantly in children and young adults from poor rural areas. It is characterized by desquamation, erythroderma, and an intense burning in the sun-exposed skin, giving rise to the term fogo selvagem (“wild fire” in Portuguese). Histopathologically and immunopathologically it is indistinguishable from pemphigus foliaceus. Unlike other pemphigus variants, this condition can be seen in multiple members of a single family. There is an increased frequency of HLA-DRB1 haplotypes, DRB1 ∗ 0404, 1402, 1406, and 1401. An environmental “second hit” is suspected but not identified. Because of the clustering of cases near rivers, Simulium black flies have been suggested as a possible vector. IgM-anti-Dsg 1 is found in a majority of fogo selvagem patients in their native environment, but is uncommon in patients with other pemphigus phenotypes and in fogo selvagem patients who move to more urban settings, further supporting a recurrent environmental antigenic exposure in the pathogenesis of this disease.
Pemphigus erythematosus (Senear–Usher’s syndrome) represents a localized variant of pemphigus foliaceus characterized by erythematous lupus-like malar dermatitis. Patients frequently manifest an abnormal antinuclear antibody test as well as the presence of pemphigus antibody. Direct immunofluorescence microscopy may show immunoglobulin and complement components along the basement membrane, as well as characteristic intercellular pemphigus antibody deposition. This disorder is believed to represent the coexistence of lupus erythematosus and pemphigus foliaceus.
IgA pemphigus comprises a recently characterized group of IgA-mediated immunobullous diseases. This entity presents as vesicopustules with neutrophils and acantholysis, most commonly affecting the axillae and groin. Oral involvement is rare. IgA pemphigus is subdivided into two histologic types: the subcorneal pustular dermatosis type with blister formation subcorneally, and the intraepidermal neutrophilic type where blisters form throughout the epidermis. The IgA pemphigus antibodies in the subcorneal pustular dermatosis type recognize desmocollin 1, whereas rare IgA pemphigus sera recognize desmoglein 1 and 3. The antigen in the intraepidermal neutrophilic form remains uncharacterized. Only 50% of patients have circulating autoantibody on indirect immunofluorescence. The subcorneal pustular dermatosis form is clinically indistinguishable from Snedden–Wilkinson’s disease and must be differentiated by immunofluorescence studies. Rarely, either form of IgA pemphigus may exhibit concomitant expression of IgG autoantibodies—so-called IgA/IgG pemphigus.
Penicillamine and angiotensin-converting enzyme (ACE) inhibitor therapies have been associated with a variety of autoimmune disorders, including pemphigus vulgaris and pemphigus foliaceus. Pemphigus foliaceus accounts for 70% of the penicillamine-induced cases, and pemphigus vulgaris composes the remainder. The development of pemphigus may occur with a wide range of dosages and is often a late complication of therapy. After discontinuation of penicillamine therapy approximately half the patients resolve in 4 months, whereas the other half require suppressive corticosteroid therapy over a longer period. Autoantibodies from drug-induced patients have the same antigenic specificity on a molecular level as do those from idiopathic forms of pemphigus.
Pemphigus has occurred in association with thymoma and myasthenia gravis. Pemphigus vulgaris, pemphigus foliaceus, and pemphigus erythematosus have all been noted associations. There is little, if any, concordance between the clinical activities of the coexistent disorders. The concurrence is believed to involve an underlying failure of thymic-dependent lymphocytes in suppressing autoimmune disease.
The description of paraneoplastic pemphigus (PNP) as a distinct disorder by Anhalt et al. focuses this issue in a small subgroup of patients who had a clear-cut association of pemphigus with a tumor. The patients are clinically heterogeneous and somewhat atypical. Clinical descriptions partially resemble Stevens–Johnson’s syndrome, with target lesions and painful oral lesions seen on occasion. Some patients have been described as having a papulosquamous eruption, whereas others have tense bullae. The most common associations, in descending order, are non-Hodgkin’s lymphoma, chronic lymphocytic leukemia, Castleman’s disease, and solid tumors including carcinomas, sarcomas, and melanoma. Histologically, suprabasilar acantholysis has been described in some cases, whereas keratinocyte necrosis, basal cell vacuolization, interface inflammation, and even basement membrane blisters have been described in others.
The immunofluorescent pattern in paraneoplastic pemphigus is one of intercellular IgG deposition, which may be patchy and focal, and basement membrane zone IgG and complement deposition. Circulating autoantibodies bind to different tissue sources than standard pemphigus antibodies, including urinary bladder, respiratory epithelium, and desmosomal areas of myocardium and skeletal muscle. Indirect immunofluorescence to rat bladder is the most specific diagnostic tool. PNP sera react to a unique complex of antigens, which includes the plakin protein family, including desmoplakin, bullous pemphigoid antigen 1 (BPAG1), envoplakin and periplakin, and desmogleins 1 and 3. Patients with paraneoplastic pemphigus may have antibodies to one or all of these antigens, or may start with a few and with time develop antibodies to other paraneoplastic pemphigus antigens. This observed progression may represent epitope spreading.
Unique to paraneoplastic pemphigus is a large proportion of patients who develop bronchiolitis obliterans, a lethal pulmonary condition characterized by severe hypoxia, a relatively clear chest X-ray, and an association in at least one patient with IgG deposition in bronchial epithelium intracellular spaces. Because desmoplakins are present in bronchial epithelium it is possible that this pulmonary finding is autoimmune-mediated. In summary, it appears that this is an unusual mucocutaneous disorder with myriad clinicopathologic findings, some of which are not a part of any of the forms of pemphigus.
Other sporadic and poorly understood associations with pemphigus include pernicious anemia, red blood cell aplasia, rheumatoid arthritis, and lymphomatoid granulomatosis.
Differential Diagnosis
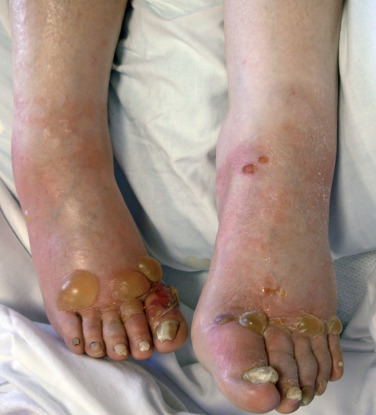
The differential diagnosis of blistering disorders of the skin ranges from a wide variety of banal dermatoses to more serious and progressive disorders such as pemphigus, bullous pemphigoid ( Fig. 16-4 ), and epidermolysis bullosa acquisita. These disorders require clinical, histopathologic, and immunopathologic evaluation for definite diagnosis. When the etiology of blistering disorders cannot be recognized, biopsy is warranted. Subsequent decisions are made on the basis of these findings. Histopathologic characteristics of potential blistering disorders that will not be discussed here in detail are listed in Table 16-1 .
|
|
|
Many patients with oral ulcerations of pemphigus are misdiagnosed as having aphthous stomatitis. After months to years, however, such patients typically progress to have extramucosal involvement.
Acantholysis is the hallmark of immunologically mediated pemphigus, but it may also be seen in Grover’s disease (transient acantholytic dermatosis), Darier’s disease (keratosis follicularis), and Hailey–Hailey’s disease (benign familial pemphigus). In Grover’s disease there is involvement of the trunk with pruritic papulovesicles, but no oral involvement. It is frequently precipitated by sun exposure or heat and lasts for weeks to months, although chronic cases are not uncommon.
Darier’s disease is an autosomal dominant disorder characterized by yellowish-brown crusted papules on the scalp, intertriginous areas, and seborrheic areas of the face and trunk. The disease is slowly progressive and seldom overtly bullous. Lesions are frequently perifollicular. Although acantholysis is suprabasilar, as in pemphigus vulgaris, characteristic dyskeratotic changes (corps ronds and grains) occur within the epidermis.
Hailey–Hailey’s disease (benign familial pemphigus) is an autosomal dominant disorder characterized by multiple grouped erythematous vesicles in intertriginous areas. Mucosal surfaces are usually spared. There is extensive loss of intercellular bridges, with partial coherence of cells throughout all levels of the epidermis.
The clinical pattern of these acantholytic disorders is usually distinctive from that of pemphigus. If confusion exists, direct immunofluorescence of perilesional skin is negative for IgG deposition in these disorders, whereas pemphigus patients demonstrate characteristic intercellular IgG deposition in stratified squamous epithelial tissues.
Patient Evaluation
Close examination of all mucous membranes is indicated. Oral involvement is the rule in pemphigus vulgaris, but esophageal as well as vulvar involvement may occur. Significant esophageal symptoms and even stricture may develop. Consequently, esophageal symptoms mandate endoscopy and possible biopsy. Patients presenting with blistering skin disorders that cannot be easily explained (e.g., friction blisters and contact dermatitis) require biopsy. If there is histopathologic acantholysis, biopsy of perilesional skin for direct immunofluorescence should be performed. Indirect immunofluorescence microscopy demonstrating pemphigus antibodies in the serum further confirms the diagnosis, and antibody titers typically correlate with disease activity. Disappearance of antibody from the serum frequently precedes remission.
Chest X-ray to rule out an associated thymoma and a search for clinical symptoms of myasthenia gravis is part of good clinical care, but a low yield of positive findings is to be expected. Culture of potentially infected lesions and close attention to protein loss and malnutrition are necessary in severe cases. Chest X-ray, tuberculosis skin test, complete blood count (CBC), and blood glucose determination should be undertaken prior to initiating corticosteroid or immunosuppressive therapy.
Treatment
Initial therapy of pemphigus involves complete suppression of blistering with oral prednisone (usually 1 to 2 mg/kg daily). Initial control with 3 to 4 mg/kg daily has been suggested, but in the authors’ estimation is associated with unnecessarily severe side effects. Because therapy may need to be continued for years, corticosteroid side effects become a major clinical problem. Corticosteroid sparing may be accomplished by the addition of an immunosuppressive agent, as discussed below. Immunosuppression is continued in sufficient doses to suppress blistering until serum antibody titers become negative, at which time tapering of therapy should be attempted. Follow-up biopsy for direct immunofluorescence once clinical manifestations have cleared for >6 months on treatment can predict the likelihood of remission once medications are stopped. Ratnam and Pang showed that three-quarters of patients with negative direct immunofluorescence at this stage remain in remission, and those with negative direct immunofluorescence who do recur have mild disease. In contrast, all patients with a positive follow-up direct immunofluorescence tend to relapse within 3 months of discontinuing therapy.
Systemic methotrexate (oral, intravenous, or intramuscular) can be used in dosages of 20 to 50 mg per week, but may aggravate oral ulcers. Cyclophosphamide is effective in oral dosages of 1 to 3 mg/kg/day, and oral azathioprine may be used in dosages of 1 to 3 mg/kg/day. Mycophenolate mofetil is effective alone or in combination with steroids in doses of 1 to 3 g/day. Close attention should be paid to a variety of potential side effects, including leukopenia, hepatotoxicity, teratogenesis, sterility, oral ulcers, and cystitis, depending on the specific agent used. Patients with excessive toxicity from oral corticosteroids or cyclophosphamide may have reduced side effects with monthly pulse doses.
Plasmapheresis may be useful in pemphigus patients poorly controlled on conventional therapy. Six-liter exchanges on three separate occasions over a 3-week period are necessary to lower the antibody titer significantly. Immunosuppressants such as cyclophosphamide are then necessary to maintain the improvement and prevent the rebound of antibody levels that usually follows plasmapheresis.
Intravenous immunoglobulin (IVIg) therapy is emerging as a promising treatment for many immune-mediated diseases. It works rapidly, and selectively lowers serum levels of pemphigus antibody. The clinical response in individual patients is variable. The concurrent administration of an immunosuppressive agent to prevent the synthesis of new antibody improves the efficacy. The dose is usually 2 g/kg/cycle, administered over 2 to 5 days. It is traditionally given in monthly cycles, but the optimal frequency of cycles is unknown. Caution needs to be taken to avoid fluid overload in elderly patients, and venous thrombosis may occur with administration.
Rituximab is a murine–human chimeric monoclonal antibody to CD20, an antigen present on B cells but not plasma cells. It is approved for use in B-cell lymphoma and is being used off-label for pemphigus and pemphigoid. Administration in a dose of 375 mg/m 2 once weekly for 4 weeks rapidly reduces the peripheral B-cell count to zero and sustains this level for 6 to 12 months. Rituximab has also been used in a dosing schedule similar to its rheumatologic use or 1000 mg initially, followed by a second infusion at 2 weeks. It seems that either dosing schedule results in a similar chance of response. Clinical improvement usually occurs within days to weeks, indicating that mechanisms other than B-cell depletion may be active. Pemphigus antibodies decrease in response to therapy and complete remission may occur. The risk of fatal infection is increased, and at present it is recommended that it be reserved for severe disease unresponsive to conventional therapy.
Because of its neutrophil-mediated pathogenesis, the drug of choice for IgA pemphigus is dapsone. Doses of 100 mg/day are usually sufficient. For patients unable to tolerate dapsone, etretinate is an alternative, providing immunosuppression by interfering with neutrophil and monocyte chemotaxis.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


