Bullous Diseases
Ilka A. Netravali
James Y.T. Wang
The skin has adhesion molecules that anchor keratinocytes to one another and to the underlying basement membrane, partly to resist mechanical trauma. Many of these molecules are targets of autoantibodies in blistering skin diseases. These autoimmune bullous diseases are divided into two major categories, depending on whether the blistering is intraepidermal (pemphigus) or subepidermal (pemphigoid). Blistering reactions also occur as a reaction to infection or drug exposure, with the most concerning presentation being that of fulminant toxic epidermal necrolysis (TEN).
While these disorders have been of the target of intense laboratory research, and mechanisms of their pathogenesis have become clearer, these conditions remain difficult to manage as their treatment necessitates the use of relatively high doses of systemic corticosteroids and immunosuppressive agents. From the perspective of a primary health care provider, these dermatoses have three unique and critical dimensions. First, they are exceedingly rare, and the diagnosis can be easily missed without a high index of suspicion. Second, access to the required diagnostic tests is readily available in the United States, and any delay in assessment and management is unjustified. This is particularly important because early diagnosis customarily results in rapid improvement and better prognosis. Third, these diseases are often better handled by specialized dermatologists with extensive experience in their diagnosis and management. The treating clinician may perform a great service by referring the patient to the closest immunodermatology expert who has a focused interest in vesiculobullous diseases.
A wide variety of autoimmune bullous dermatoses exists, but this section will focus on the two most etiologically relevant members of the pemphigus and pemphigoid classes of diseases, respectively: pemphigus vulgaris and bullous pemphigoid. erythema multiforme and the Stevens-Johnson syndrome/toxic epidermal necrolysis spectrum will also be covered in separate sections.
Pemphigus Vulgaris
 |
A 49-year-old white man was referred to a dermatologist with a 5-month history of a bullous disorder. The patient initially presented to his internist with gingival and buccal erosions; occasional flaccid blisters, crusted papules, and plaques involving the scalp, back, face, and chest were also noted (Fig. 23-1). A diagnosis of pemphigus vulgaris was made when indirect immunofluorescence testing revealed IgG deposits on the surface of keratinocytes. The patient initially failed to respond to systemic prednisone and azathioprine but ultimately responded to cyclophosphamide. Metastatic carcinoma (primary unknown) was diagnosed 6 months later (approximately 18 months after presentation) and, despite chemotherapy, the patient died.
Background
Pemphigus comprises a group of chronic, progressive autoimmune diseases that cause blistering and ulceration of the skin, mucous membranes, or both, loss of normal epithelial cell–cell adhesion (acantholysis), and the presence of pathogenic IgG autoantibodies directed against transmembrane desmosomal proteins termed desmogleins. There are two basic forms of pemphigus—vulgaris and foliaceus—that affect different layers of the skin, have unique symptoms, and target distinct antigens. In the “deep” pemphigus vulgaris, blisters develop just above the basal-cell layer and are associated with autoantibodies to desmoglein 3, a keratinocyte cell-surface adhesion molecule. In the “superficial” pemphigus foliaceus, the blisters are high in the epidermis, just below the stratum corneum, and are associated with antibodies against desmoglein 1, another cell-surface adhesion molecule. Several subtypes of each form exist.
Key Features
Epidemiology: rare; equal in men and women; onset 40 to 60 years of age; more common in Jewish or Mediterranean descendants.
Clinical features:
Generalized flaccid blisters or crushed erosions, located on head, upper trunk, intertriginous areas, and mucosa; often begins on oral mucosa, may lead to hoarseness; skin is usually painful and Nikolsky sign is positive.
Extracutaneous manifestations: oral mucosal erosions; rarely, thymoma and myasthenia gravis.
Caused by autoantigens: desmoglein 3.
Diagnosis:
Light microscopy: supra-basilar blister with acantholysis and vesicle formation, eosinophilic infiltrate.
Direct immunofluore-scence: intercellular IgG and/or C3 on keratinocyte cell surface.
Indirect immunofluore-scence: IgG against keratinocyte cell surface.
Therapy: Corticosteroids (prednisone 1 mg/kg/d) taper to maintenance; may add azathioprine, methotrexate, cyclophosphamide, mycophenolate mofetil, plasmapheresis.
Note: May be fatal if untreated; risk of infection in treated and untreated cases.
Epidemiology
Pemphigus is a rare disease with an annual incidence of 0.75 to 5 cases per million. Incidence of the different forms, however, varies across the globe: pemphigus vulgaris (PV) is most common in the United States and Europe. PV can develop at any age, but is typically diagnosed in the fourth to sixth decades of life. The disease can affect anyone, occurring in equal frequency for both sexes; however, it is most prevalent in those of Mediterranean or Jewish descent. Individuals with certain human leukocyte antigen (HLA) allotypes are predisposed to the disease, though the susceptibility gene varies according to ethnic origin. PV rarely affects more than one family member and patients are of various HLA types; it is therefore not considered a hereditary disease.
Pemphigus foliaceus (PF) is more prevalent in Africa and in certain rural areas in underdeveloped nations, where it afflicts up to 3% of the population. Sporadic and endemic forms of PF exist. The sporadic form is most frequent in the United States and Europe, where its incidence ranges from 0.1 to 0.2 of PV. Endemic PF (also termed fogo selvagem and Brazilian PF) is a variant that is usually diagnosed in certain regions of Brazil, Tunisia, and Colombia. Clinically and histologically, the disease is similar to sporadic PF. The endemic form, however, typically afflicts individuals in late adolescence and early adulthood. Importantly, it often affects multiple members of the same family. The epidemiology of the endemic form suggests an environmental cause, which remains unidentified, although an insect vector is suspected. Endemic PF typically arises at the border between developing and undeveloped areas; as an area becomes industrialized, the disease disappears.
PV and PF are associated with myasthenia gravis and thymic abnormalities, including benign or malignant thymomas and thymic hyperplasia. Although irradiation of the thymus or thymectomy is helpful in management of myasthenia gravis, they do not necessarily reduce pemphigus disease activity.
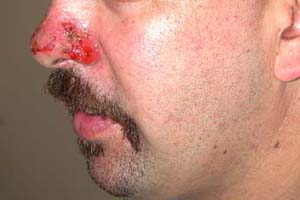
 Figure 23-1 Pemphigus vulgaris on the nose. There are no remaining bullae, only erosion and crust (Photo courtesy of Lynne Morrison, MD). |
Clinical Presentation
Pemphigus Vulgaris (“Deep Pemphigus”)
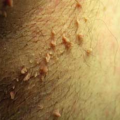
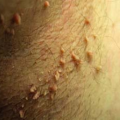
Virtually all patients with this deep form of pemphigus first present with painful, nonhealing ulcerations in the mouth. Blisters are rare (Fig. 23-2), as they rupture soon after forming to leave an ulcerated area (Fig. 23-3). Ulcerations are usually multiple, superficial, and irregular in shape, and arise from mucosa of healthy appearance. Although any surface can be involved, the most common sites are the buccal and labial mucosa, the palate, and the tongue (Fig. 23-4). The ulcers of PV do not heal, in contrast to the oral lesions of aphthous stomatitis or viral infections that heal over days to weeks. Ulcers are also multiple, which differentiates them from an ulcerated tumor, which is single.
Because the disease is rare, PV is not often diagnosed at a first examination, but rather considered only after lesions have been present for weeks to months and the patient has not responded to antibiotic, antifungal, or antiviral therapy. PV should be considered in anyone who has multiple, nonhealing oral ulcers that persist for longer than a month.
 Figure 23-2 An intact flaccid bulla of pemphigus vulgaris. From Berg D, Worzala K. Atlas of Adult Physical Diagnosis. Philadelphia: Lippincott Williams & Wilkins, 2006. |
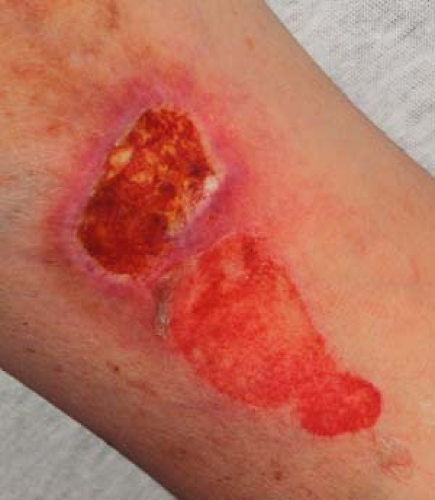 Figure 23-3 Erosions and ulcerations from pemphigus vulgaris. From Berg D, Worzala K. Atlas of Adult Physical Diagnosis. Philadelphia: Lippincott Williams & Wilkins, 2006. |
 Figure 23-4 Oral erosion on the posterior palate from pemphigus vulgaris. |
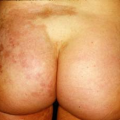
 Figure 23-5 Pemphigus foliaceus on the chest. |
The condition progresses over weeks to months with lesions developing on the skin and symptoms indicating nasal and esophageal involvement. Occasionally, such skin lesions are the initial manifestations. They usually begin as small blisters filled with a clear fluid, arising from normal-appearing skin. The blisters are typically flaccid because the overlying epidermis is thin and cannot sustain much pressure. Because they are thin, the blisters rupture in several days, and are replaced by well-demarcated, coin-sized, superficial erosions with a surrounding collar of loose epidermis (Fig. 23-1). Lesions appear most often on the scalp, upper chest, and back and have predilection for the medial portion of the torso rather than the sides. The face and neck are also commonly involved, but any surface covered by stratified squamous epithelium can be affected.
Sites that are often missed on examination include the periungual areas; the pharynx and larynx, indicated by pain on swallowing food and by hoarseness; the nasal cavity, manifested by nasal congestion and morning mucous discharge; and the cervix.
Nikolsky sign: a characteristic mechanical feature of all forms of active and severe pemphigus is the Nikolsky sign, in which firm, sliding pressure on normal-appearing skin causes separation of the epidermis from the underlying dermis. This sign is elicited most easily adjacent to an active lesion.
Pemphigus Foliaceus (“Superficial Pemphigus”)
In this superficial form of pemphigus, there is insufficient overlying tissue to trap fluid and cause blister formation. Lesions typically start with multiple, pruritic, crusted, coin-sized patches on the upper torso, face, and scalp (Fig. 23-5). They arise from healthy-looking skin and are likened to cornflakes. The crusts can be removed easily, leaving superficial erosions. Untreated lesions do not heal, and increase in number over weeks to months. Lesions can become confluent to resemble exfoliative dermatitis in severe cases. Notably, oral involvement in PF is rare, in contrast to that seen in deep PV.
Drug-induced Pemphigus (DIP)
Certain drugs can trigger both PV and PF (Table 23-1). Though uncommon, this possibility should be excluded in all patients with newly diagnosed disease.
The clinical, histological, and immunofluorescence abnormalities of drug-induced and idiopathic pemphigus are similar. The most commonly implicated drugs contain sulfhydryl groups and include penicillamine and the angiotensin-converting enzyme inhibitors, such as captopril. In contrast to other drug-induced eruptions, DIP may not develop for up to several months after initiation of treatment with the offending agent. Most patients with DIP have a good prognosis and clear with adequate therapy and discontinuing of the causative agent.
The clinical, histological, and immunofluorescence abnormalities of drug-induced and idiopathic pemphigus are similar. The most commonly implicated drugs contain sulfhydryl groups and include penicillamine and the angiotensin-converting enzyme inhibitors, such as captopril. In contrast to other drug-induced eruptions, DIP may not develop for up to several months after initiation of treatment with the offending agent. Most patients with DIP have a good prognosis and clear with adequate therapy and discontinuing of the causative agent.
Table 23-1 Medications Implicated in Pemphigus | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Pathogenesis
The basic abnormality in all forms of pemphigus is acantholysis, or the separation of keratinocytes from one another. The primary event is the dissolution of the intercellular substance (ICS), followed by separation of desmosomes. This process leads to the formation of a cleft within the epidermis, which subsequently enlarges into a bulla. All forms of pemphigus are characterized by circulating and skin-fixed autoantibodies, referred to as intercellular antibodies, against keratinocyte cell-surface antigens. Circulating intercellular antibodies are present in about 80% of patients with active disease, and their titers usually correlate with disease activity. Tissue-fixed intercellular antibodies are present in lesions and in adjacent healthy skin in about 90% of patients. They are usually IgG, although IgM, IgA, and the complement protein C3 may also be deposited.
Intercellular antibodies are directed against multiple keratinocyte cell-surface antigens, of which the adhesion molecules, desmogleins 1 and 3, are best characterized. The antigens targeted in the two forms of pemphigus differ. In PV, intercellular antibodies are predominantly directed against desmoglein 3 and less often against desmoglein 1; in PF, they are predominantly directed against desmoglein 1.
Scant knowledge exists regarding the generation of the intercellular antibodies. The disease is idiopathic in most individuals, although it is triggered in some by an external cause, such as a drug (Table 23-1). Different drugs cause different forms of pemphigus. Many of the frequently associated medications contain sulfhydryl groups that may interact with sulfhydryl groups found in desmogleins 1 and 3 to increase the antigenicity
of these adhesion molecules. Importantly, significant inflammatory cell infiltrate is not observed in pemphigus lesions, suggesting that cellular immune mechanisms are not directly involved in the pathogenesis. It remains possible, however, that T-cell responses play a role in regulating the formation of the pathogenic autoantibodies.
of these adhesion molecules. Importantly, significant inflammatory cell infiltrate is not observed in pemphigus lesions, suggesting that cellular immune mechanisms are not directly involved in the pathogenesis. It remains possible, however, that T-cell responses play a role in regulating the formation of the pathogenic autoantibodies.
Diagnosis
Diagnostic Methods
Diagnosis is based on three independent sets of criteria: clinical features, histology, and immunologic tests.
Clinical Features
PV: classic findings include multiple flaccid blisters arising from healthy skin that have a tendency to extend at their periphery, multiple chronic oral ulcers, and a positive Nikolsky sign.
PF: distinguishing characteristics include easily rupturing blisters leaving superficial erosions that are usually confined to the face, neck, and upper trunk.
Histology
PV: intraepidermal blisters characterized by suprabasal acantholysis of keratinocytes.
PF: acantholytic skin cleavage occurs just at the level of the stratum granulosum.
Immunologic tests
These assays are used to detect the circulating and tissue-fixed intercellular antibodies against keratinocyte surface antigens that are encountered in all forms of pemphigus.
Indirect immunofluorescence (IIF) and direct immunofluorescence (DIF): circulating intercellular antibodies are detected by IIF assays on patient serum (with monkey esophagus serving as an ideal substrate for PV and guinea pig for PF). Tissue-fixed intercellular antibodies are identified by DIF assays on skin biopsies.
Enzyme-linked immunosorbent assay (ELISA): ELISA is also available to detect antibodies to desmoglein 1 and desmoglein 3. The presence of antibodies against desmoglein 3, sometimes together with those against desmoglein 1, is associated with PV, whereas antibodies to desmoglein 1 alone are associated with PF. ELISA is more specific and somewhat more sensitive than IIF.
The presence and concentration of the intercellular autoantibodies relate to disease activity. Increases in titers may precede clinical flares, and they usually disappear in patients in remission.
Differential Diagnosis
Pemphigus must be differentiated from the many other conditions that also cause skin blisters or oral ulcers and erosions (Table 23-2) (Figs. 23-6 to 23-10). Blisters arise as a sign of many other autoimmune, genetic, and metabolic diseases:
Blisters in most of these cases occur within or below the dermal-epidermal junction (DEJ) and can be readily excluded by skin biopsy.
Biopsy: for maximal information, a deep shave or 4-mm punch biopsy should be performed for routine light microscopy at the edge of an active
lesion. Adjacent healthy skin should also be included so that the level of the blister can be accurately identified.
Table 23-2 Differential Diagnosis of Pemphigus
INTRAEPIDERMAL BLISTERING DISEASES WITHOUT AUTOANTIBODIES
- Familial benign pemphigus (Hailey-Hailey disease) (Fig. 23-6)
- Bullous impetigo, staphylococcal scalded skin syndrome
- Blisters of herpes simplex and zoster
- Allergic contact dermatitis (Fig. 23-7)
- Epidermolysis bullosa simplex
- Incontinentia pigmenti
- Aphthous ulcers
- Candidiasis
- Behçet disease
- Bullous pemphigoid
- Herpes gestationis
- Cicatricial pemphigoid (Figs. 23-8, 23-9)
- Epidermolysis bullosa acquisita
- Linear IgA disease and chronic bullous disease of childhood
- Dermatitis herpetiformis (Fig. 23-10)
- Bullous lupus erythematosus
- Erythema multiforme
- Toxic epidermal necrolysis
- Porphyria
- Epidermolysis bullosa

Figure 23-6 Hailey-Hailey disease in the axilla.
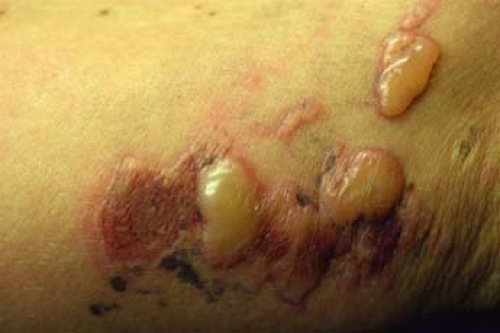
Figure 23-7 Bullous allergic contact dermatitis from poison ivy.- Familial benign pemphigus (Hailey-Hailey disease) (Fig. 23-6)
Paraneoplastic Pemphigus (PNP)
PNP is sometimes judged as a variant of pemphigus because it is associated with acantholysis and with antibodies against desmoglein 1 and 3; it is usually associated with an occult lymphoproliferative neoplasm (especially B-cell lymphoma/leukemias, thymomas, and Castleman tumors), however, and may be considered a separate disease:
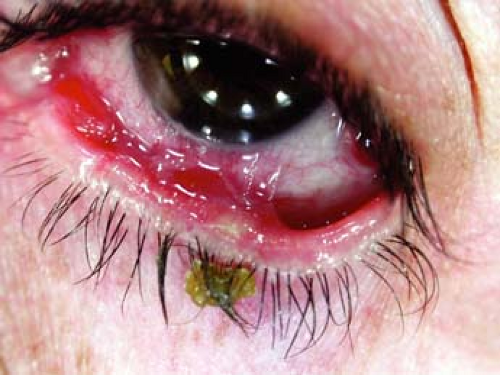 Figure 23-8 Cicatricial pemphigoid. Symblepharon formation. |
As with PV, the skin lesions in PNP can arise both in the oral mucosa and skin, and there is histologic acantholysis and intercellular antibodies on both DIF and IIF.
PNP has certain features that differentiate it from other forms of pemphigus:
Clinical: extensive, recalcitrant oral lesions characteristically involve the vermillion border of the lips and the polymorphic skin lesions often resemble those of erythema multiforme or toxic epidermal necrolysis.
Histologic: in addition to suprabasal clefting and acantholysis, interface changes (i.e., inflammation) occur at the DEJ.
Immunologic: on DIF, abnormal deposits of Ig, complement, or both, are also present at the DEJ (in addition to on the surfaces of keratinocytes). Intercellular antibodies also react against columnar and transitional epithelia. Most importantly, the antibodies are also directed against plakin proteins (e.g., envoplakin and periplakin). None of these abnormalities occur in pemphigus.
 Figure 23-9 Oral cicatricial pemphigoid. Note intact bulla in posterior pharynx. |
This disease is generally progressive and often fatal within 2 years if associated with a malignant neoplasm. Treatment of the underlying malignancy does not influence the skin disease, although treatment of associated benign neoplasms does lead to resolution of PNP.
Treatment
Treatment modalities used to control pemphigus are categorized into those that act rapidly, and those with a delayed effect that are used in chronic management to reduce the requirement for systemic corticosteroids. The goal of treatment is to achieve a complete remission with the least side effects. The three phases of treatment are control, consolidation, and maintenance.
 Figure 23-10 Dermatitis herpetiformis. Intact bullae and erosions/ulcerations from scratching on the buttocks. |
Control phase: in this period, treatment is swiftly intensified until disease activity is suppressed, as confirmed by a major reduction of new lesion formation, lack of itching, and the start of healing of established lesions. Pemphigus typically responds to treatment within 2 weeks if the correct dose of medication is used; continued disease activity implies insufficient treatment or a complicating factor.
The initial treatment is chosen according to the extent and rate of progression of lesions. If lesions are few and progressing gradually, they can be treated individually with topical, high-potency corticosteroid ointments or with intralesional injections of corticosteroids. New lesions that incessantly appear at rapid rates should be managed with a low dose of oral prednisone (20 to 40 mg/d).
Patients who do not respond, or have initially extensive disease are treated with a moderately high dose (70 to 90 mg/d). The dose is increased in 50% increments every 1 to 2 weeks until disease activity is controlled. Importantly, if only corticosteroids are used, this dose may rise to over 240 mg/d in resistant cases.
If a response is not seen with prednisone at 120 mg/d or more, other treatment options should be attempted, such as plasmapheresis, intravenous immunoglobulins (IVIg), or intravenous methylprednisolone at 1 g/d for 5 days. If no improvement is observed to any of the presented options, there may be secondary bacterial, fungal, or viral (herpes simplex) contamination of skin lesions, which must be treated. Appropriate culture of skin lesions can exclude this possibility.
Consolidation phase: the objective of this phase is to maintain the type and dose of treatment(s) needed to control disease activity until the majority of lesions have healed. Like the control phase, this period usually lasts for weeks. If lesions are observed to heal slowly, the strength of therapy is inadequate, and needs to be increased. To prevent future flares, medications should not be tapered until about 80% of lesions have healed.
Maintenance phase: this period begins when most lesions have healed and represents the gradual tapering of medication doses to the lowest levels required to prevent the appearance of new lesions. The long-term goal here is to ultimately discontinue all systemic treatments and this can be achieved in most patients. If multiple drugs are being used, these should be tapered one at a time. Importantly, tapering too rapidly will increase the chance of a flare, while tapering too slowly may lead to unnecessary side effects.
Adjuvant therapies: although systemic corticosteroids are the mainline treatment for pemphigus, the side effects that are associated with the required high and prolonged doses can be severe. Adjuvant therapies should be considered in the presence of contraindications to the use of systemic corticosteroids, development of serious side effects to corticosteroids, or the inability to reduce corticosteroid doses without repeated flares in disease activity.
Consider immunosuppressive such as cyclophosphamide, azathioprine, cyclosporine, methotrexate, and mycophenolate mofetil, anti-inflammatory drugs including gold, dapsone, and antimalarials, and antibiotics such as tetracycline and minocycline.
Most of these agents are characterized by a 4- to 6-week lag phase before becoming effective.
Topical treatments: these modalities are used to alleviate pain and prevent and treat secondary infections, which can delay the response to therapy.
Pain and the sticking of clothing to lesions can be lessened with petrolatum-based ointments.
Normal saline compresses or diluted bacteriostatic solutions, such as silver nitrate or potassium permanganate, can be used to keep lesions clean.
Stubborn lesions can be treated with topical high-potency corticosteroids like clobetasol, or with intralesional corticosteroid injections. Intralesional corticosteroids are more successful in high doses (e.g., 20 μg/L triamcinolone acetonide).
Unresponsive lesions should be cultured to exclude secondary infection.
Oral lesions: oral lesions respond significantly slower to therapy than their skin counterparts and as such, pose a treatment challenge. Successful strategies include the use of high-potency topical corticosteroids such as clobetasol and steroids in an adherent base such as triamcinolone acetonide in gelatin; allowing corticosteroid tablets to dissolve in the oral cavity rather than swallowing, and rinsing with dexamethasone solution may also be effective. The best results, however, are achieved with intralesional injections of triamcinolone acetonide at 20 μg/L. Secondary candidiasis commonly occurs, but is managed with antifungals. Laryngoscopy should be employed to evaluate throat pain on swallowing to differentiate pemphigus and secondary candidiasis. As emphasized above, patients should be followed closely and regular monitoring of side effects should continue throughout therapy duration.
Prevention of Disease Flares
Many factors that aggravate pemphigus should be avoided. These include dental work, sun exposure, radiographs, stress, and trauma. Disease flares following dental work are common enough to merit prophylactic administration of 20 mg/d prednisone (in addition to the patient’s normal requirement) for 5 to 7 days, for each dental procedure associated with trauma to the gums. Patients should also be instructed to avoid unnecessary exposure to the sun and to use broad-spectrum sun-protective creams and clothing.
Course and Complications
Pemphigus Vulgaris (PV)
The clinical course of PV is rather unpredictable, but invariably chronic. Several factors direct the clinical outcome: age of onset, severity of disease, response to systemic corticosteroids, and disease progression. Typically, however, bullae and erosions will spread if left untreated (Fig. 23-1). As with burns, these lesions when widespread can be complicated by metabolic disturbances or severe infection and sepsis, which are leading causes of death. Before the advent of systemic corticosteroids, about 75% of patients who developed PV died within a year. With treatment, however, lesions heal with crusting followed by re-epithelialization. There is no scarring, although transient residual hyperpigmentation can occur at sites of former lesions. Most patients with PV eventually enter a phase of partial remission in which they can be maintained, lesion-free, with minimum (<15 mg/d prednisone) doses of corticosteroids, or achieve complete remission in which they are lesion-free and require no therapy. Unfortunately, the immunosuppressive therapy is also a major contributor to the development of infections of the denuded epithelium and the resultant fatal sepsis that often claims the lives of PV patients.
Referral to a dermatologist, especially one interested in bullous disorders, may be helpful in initial workup and management.
Consider ear, nose, and throat examination if pharyngeal or esophageal involvement is present.
Ophthalmologic consultation may be helpful for ocular disease and topical therapy.
Secondary HSV or bacterial infection.
Consider the patient’s medications as a potential cause and stop potentially offending families of drugs.
Pemphigus Foliaceus (PF)
The prognosis of untreated PF is better than that of PV, mostly due to the superficial nature of the lesions, and because of the lower risk of infection, fluid loss, and metabolic disturbance. The treatment, however, is no easier, because the doses of drugs needed to control PF are similar to those used for PV.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


