Abstract
Autoinflammatory syndromes are a recently described group of disorders characterized by inflammation mediated by the innate immune system. All these syndromes have cutaneous presentations in addition to multiorgan involvement. The first syndromes recognized have been monogenic with mutations in genes predominantly allowing dysregulation of the interleukin-1 pathway. More recently autoinflammatory-like processes have been identified in more common diseases such as acne and psoriasis.
Keywords
Autoinflammation, periodic fever syndromes, interleukin 1
- •
Autoinflammatory syndromes are characterized by excessive multisystem inflammation from activation of innate pathways rather than the acquired immune system.
- •
Autoinflammatory syndromes are mostly rare monogenic disorders that generally first present in childhood.
- •
Most of these diseases are, at least in part, modulated by the interleukin-1 pathway, which has provided targeted treatment strategies. Tumor necrosis factor-alpha antagonists have also proved useful agents for these diseases.
Introduction
Inflammation is a highly adapted necessary response to both infection and neoplasia. However, damage to tissues may occur from maladaptive autoimmune processes where self-directed T-cells or antibody-producing B-cells are activated in disorders such as systemic lupus erythematosus. Alternatively, damage may be initiated by various germline-encoded innate immune receptors with the absence of involvement of the adaptive immune system. The term “autoinflammation” has been coined for this process. This chapter will outline the various syndromes where dysregulation of innate immunity is central to their pathogenesis.
Molecular Primer
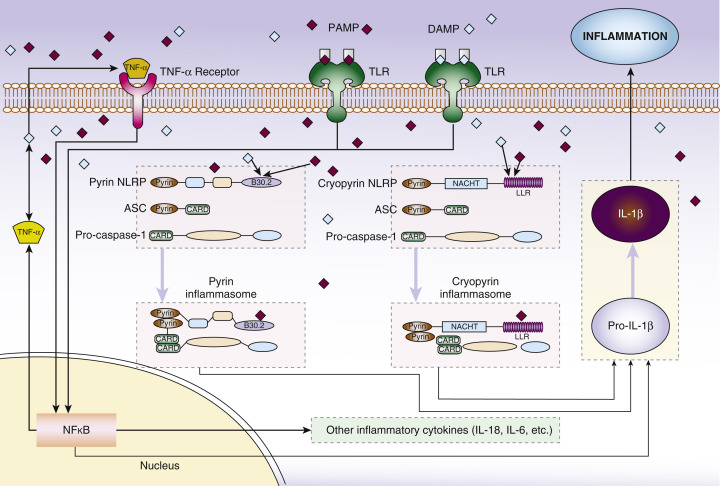
Various molecules that are recognized by cells of the innate immune system mediate the initiation of host response to infection or tissue damage ( Fig. 8-1 ). Chiefly, these are called pathogen-associated molecular patterns (PAMPs), which recognize bacterial and viral components, and danger-associated molecular patterns (DAMPs), which recognize signals related to cell death and toxins. PAMPs and DAMPs interact with pattern recognition receptors such as Toll-like receptors and Nod-like receptors (NLRs), which leads to downstream inflammation. This inflammation is predominantly mediated by the relatively recently described multimeric proteins called inflammasomes. Most autoinflammatory syndromes have genetic mutations in these pathways that lead to increased and inappropriate inflammasome activity ( Table 8-1 ).
| Cryopyrin Associated Periodic Syndrome (CAPS) | Schnitzler’s Syndrome (SS) | Deficiency of IL-1 Receptor Antagonist (DIRA) | Tumor Necrosis Factor Receptor Associated Periodic Syndrome (TRAPS) | Familial Mediterranean Fever (FMF) | Hyperimmunoglobulinemia D Syndrome (HIDS) | Pyogenic Arthritis, Pyoderma Gangrenosum and Acne Syndrome (PAPA) | Synovitis, Acne, Pustulosis, Hyperostosis and Osteitis Syndrome (SAPHO) | |
|---|---|---|---|---|---|---|---|---|
| Cutaneous findings | Daily nonitchy urticarial rash | Urticarial rash | Pustular rash, Ichthyosis | Erythema overlying areas of myalgia, Periorbital edema | Erysipelas-like erythema | Exanthem-like rash, urticarial and purpuric lesions also reported | Acne, pyoderma gangrenosum | Acne, palmoplantar pustulosis |
| Systemic features | Arthralgia, sensorineural healing loss, neurological impairment, systemic amyloidosis | IgM paraprotein, arthralgia/myalgia, bone pain | Fetal distress, osteolytic lesions, periostitis, epiphyseal ballooning, multiorgan failure | Periodic febrile episodes, myalgia, abdominal cramping and nausea | Febrile episodes, peritonitis, pleuritis, synovitis, systemic amyloidosis | Febrile episodes, abdominal pain, arthralgia, lymphadenopathy, splenomegaly | Monoarthritis typically seen in first decade, cutaneous manifestations present later (puberty) | Synovitis, osteoarticular disease affecting chest wall, sacroileitis |
| Age of onset | Infancy | >40 years (typically) | Neonatal | 3 years (rarely present in adulthood) | <20 years | Infancy | <10 years | Any |
| Inheritance | AD | N/A | AR | AD | AR | AR | AD | Unknown |
| Gene | NLR3P | N/A | IL1RN | TNFRSF1A | MEFV | MVK | PSTPIP1 | N/A |
| Protein | Cryopyrin | N/A | IL-1 receptor antagonist | TNFRSF1A | Pyrin | Mevalonate kinase | CD2 binding protein | N/A |
| Treatments | Anakinra, rilonacept, canakinumab | Colchicine, NSAIDs, anakinra | Anakinra | Corticosteroids during attack, etanercept, anakinra, canakinumab | Colchicine, thalidomide, sulfasalazine, TNF blockers, anakinra | Simvastatin, corticosteroids, etanercept, anakinra | Etanercept, adalimumab, infliximab, anakinra, corticosteroids, retinoids for acne | NSAIDs and intra-articular corticosteroids for joint disease, methotrexate, azathioprine, TNF blockers, anakinra |
Cryopyrin-Associated Periodic Syndrome
Cryopyrin-associated periodic syndrome (CAPS) is the preferred diagnostic term for three originally separately described syndromes: familial cold autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS), and neonatal onset multisystem inflammatory disorder (NOMID). In practice, the severity of clinical presentation is observed as mild (FCAS), moderate (MWS), and severe phenotypes.
Pathogenesis
CAPS is an autosomal dominant inherited condition. It is caused by “gain-of-function” mutations in NLRP3 , about half of patients have a family history of CAPS with the remainder representing spontaneous (new) mutations. NLRP3 encodes cryopyrin, which activates the NLRP3 inflammasome. The downstream effect of this is excessive production of the proinflammatory cytokine interleukin-1 (IL-1), which is responsible for the clinical manifestations of CAPS.
Clinical Features
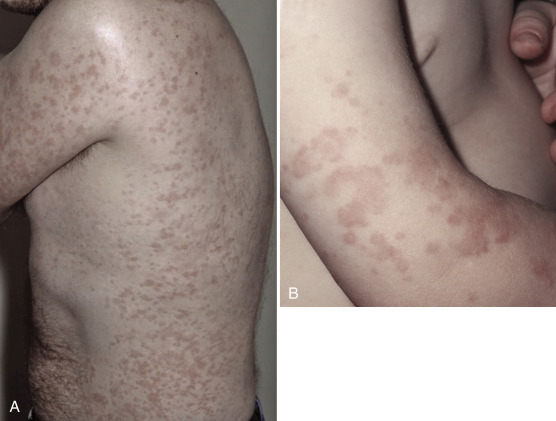
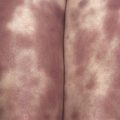
Characteristically, patients present with an urticarial rash in the neonatal period ( Fig. 8-2 ). This differs from chronic urticaria in a number of ways. The rash is present lifelong, tends not to be pruritic although patients may describe heat or tightness in affected skin, and has a circadian rhythm with little or no rash in the morning with progressive worsening as the day progresses. It is associated with constitutional symptoms, which include fever, arthralgia, headache, myalgia, and conjunctival injection. Most patients will describe these inflammatory episodes on a daily basis and furthermore may be precipitated or worsened by other factors like cold exposure. As the clinical severity advances patients may additionally have high-tone sensorineural hearing loss and systemic amyloidosis. The most severely affected patients may have neurological symptoms from raised intracranial pressure resulting in neurological impairment and rapidly progressive deforming arthropathies.
Evaluation
Skin biopsy is helpful, as it tends to reveal a neutrophilic dermal infiltrate, which is perieccrine and perivascular without evidence of vasculitis. Genetic evaluation may reveal a germ-line mutation in NLRP3 , although a significant proportion of patients will have negative mutational analysis on conventional genetic testing. It has been shown recently that the majority of these patients are actually mosaic for NLRP3 mutations. Acute phase reactants such as C-reactive protein (CRP) will be elevated, as will serum amyloid A (SAA) although this lab test is not widely available in clinical practice. Due to prolonged elevation of SAA, patients are susceptible to systemic amyloidosis (AA type). The most serious sequelae of amyloidosis is the development of renal impairment secondary to nephrotic syndrome; therefore, regular urinalysis is required for the monitoring of proteinuria. Audiometry should be performed at initial assessment to identify sensorineural hearing impairment. Referral to rheumatology and neurology may also be considered.
Treatment
Drugs that block the IL-1 pathway are highly efficacious and generally achieve complete clinical remission for patients. There are currently three drugs that the Food and Drug Administration has approved for use in CAPS. Anakinra, which is an IL-1 receptor antagonist; daily dosing is required as its half-life is 4 to 6 hours. Rilonacept is an IL-1 trap molecule with a longer half-life so requires weekly administration. Canakinumab is a fully human monoclonal antibody directed against IL-1β and is dosed every 8 weeks. This class of drugs is generally well tolerated but side effects include rare neutropenia and increased risk of infection.
Schnitzler’s Syndrome
In the 1970s, Liliane Schnitzler, a French dermatologist, reported a syndrome characterized by chronic urticaria, bone pain, fever, and the presence of an IgM paraprotein. Since the original observations about 200 cases have been reported in the literature.
Pathogenesis
Although IgM or rarely IgG gammopathy is the invariable biological characteristic of the disease, it is unclear how this contributes to the pathophysiology. It is evident that the cytokine IL-1β is elevated and contributes to the clinical features of systemic inflammation. The effects of IL-1β on the gammopathy or vice versa are unknown.
Clinical Features
Schnitzler’s syndrome (SS) should be suspected in any patient with a chronic recurrent urticarial rash, fever, and arthralgia/myalgia or bone pain. The urticarial lesions are similar to patients with CAPS or adult-onset Still’s disease (AOSD) as they are evanescent and rarely itchy. Additionally, patients may have lymphadenopathy and hepatosplenomegaly. Phenotypically there is a broad overlap between SS, CAPS, and AOSD, which is likely explained as they all result in elevated levels of IL-1β. A feature that differentiates them is age of onset; SS tends to present after the age of 40 whereas CAPS is typically lifelong. Still’s disease may present initially with a severe pharyngitis.
Evaluation
Serum electrophoresis and/or immunofixation are key to establishing a diagnosis of SS. Other laboratory makers include a leukocytosis and raised inflammatory markers including erythrocyte sedimentation rate (ESR) and CRP. Transaminitis is rarely seen in SS but is common in AOSD, as is a very elevated ferritin level. Skin histology will reveal a neutrophilic infiltrate but no evidence of leukocytoclastic vasculitis as would be evident in urticarial vasculitis. Regular monitoring of the gammopathy is important as approximately 20% of patients will develop a lymphoproliferative disorder, which is similar in proportion to that seen in other patients with monoclonal gammopathy of uncertain significance.
Treatment
In patients with mild disease, a therapeutic trial of colchicine is reasonable. Nonsteroidal anti-inflammatory drugs (NSAIDs), such as ibuprofen, may help with bone or joint pain. There are reports of hydroxychloroquine being helpful for a small number of patients. In patients with significant impairment of quality of life and/or persistent raised inflammatory markers, anakinra can be commenced, which is generally highly effective.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


