Key points
- •
Atopy of the skin is manifested as atopic dermatitis (atopic eczema).
- •
Histologically, the atopy of the skin is characterized by epidermal and dermal infiltration predominantly by lymphocytic cells as well as eosinophils and mast cells. In some patients, the granular cell layer is diminished because of the absence of filaggrin protein.
- •
The skin surface of patients affected by skin atopy is commonly characterized by lack of proper hydration (xerosis), and some patients have a genetic mutation of a key skin barrier component, filaggrin. We frequently observe pathogenic Staphylococcus colonization, with a relative decrease in commensal bacteria and overall microbial diversity, a phenomenon known as dysbiosis.
- •
Physiologically, patients affected by skin atopy bear heavy burdens of itch and pain, which in turn affects patients’ sleep, circadian rhythm, and overall health. Severe disease associates with cardiovascular and cerebrovascular comorbidities.
- •
On the immunologic aspect, patients affected by skin atopy typically have deviations of both innate and adaptive systems. The documented findings include deficiency in antimicrobial peptides and helper T-cell imbalance that favors Th2 with upregulation on the expressions of interleukin-4 (IL4), IL5, IL10, IL13, IL31, and serum immunoglobulin E (IgE).
Introduction
When atopy occurs in the skin, the medical term is atopic dermatitis or atopic eczema . In this chapter, a bird’s-eye view on major aspects of atopic dermatitis will be discussed, including the histology of inflammatory skin lesion, the skin surface abnormalities, the physiologic abnormalities of atopic dermatitis, and the altered immune milieu. The chapters that follow will expand the overview of atopic dermatitis with detailed delineation. This chapter, in fact, is the miniature representation of this entire book on clinical and basic sciences of atopic dermatitis.
Atopy of the skin under the microscope: histopathology
Historically, one of the earliest attempts by the medical community to understand the pathomechanism of disease is through a microscopic examination of disease tissue (histopathology), as it is an identification of what the disease is and what damages the disease causes at the tissue level. The basic process involves getting the disease tissue, preserving the tissue with formaldehyde (formalin), dehydrating the tissue in graded alcohol, embedding and hardening the tissue in paraffin, sectioning and placing thin sections of tissue on glass slides, rehydrating the tissue, and staining the tissue sections with routine combination stain of hematoxylin (for cell nucleus) and eosin (for cellular cytoplasm). After mounting the stained tissues with a coverslip, the tissues on slides can then be examined microscopically. Although not particularly high-tech compared to newer diagnostic methods, histopathology provides very important information regarding a particular disease, including tissues and level affected, structures and/or cells damaged, invading pathogens detected, inflammatory cells involved, cancerous cell type identified, and boundary of the cancerous tissue delineated. When needed, special stains other than hematoxylin/eosin are used to detect other components with better visualization. Giemsa stain is used to identify mast cells, periodic acid–Schiff (PAS) stain detects fungal elements, Gram stain reveals bacteria, Warthin-Starry stain labels spirochetes bacteria, and acid-fast stain identifies mycobacteria ( ). Besides these special staining techniques that utilize chemical stains, immunohistochemistry staining methods use antibody to identify certain cellular components for visualization after the sectioned tissues undergo certain antigen retrieval steps. These retrieval steps are necessary since formalin fixation disguises the antigenic epitope, and the unmasking steps are required to make these antigenic epitopes available for the antibody binding ( ). Tissues obtained for regular histopathology can also be specially preserved and processed for electron microscopic examination if indications for the more detailed examination arise ( ). We can apply molecular diagnostic methods to the tissue sections processed through the routine formalin-fixed paraffinized tissues as well ( ).
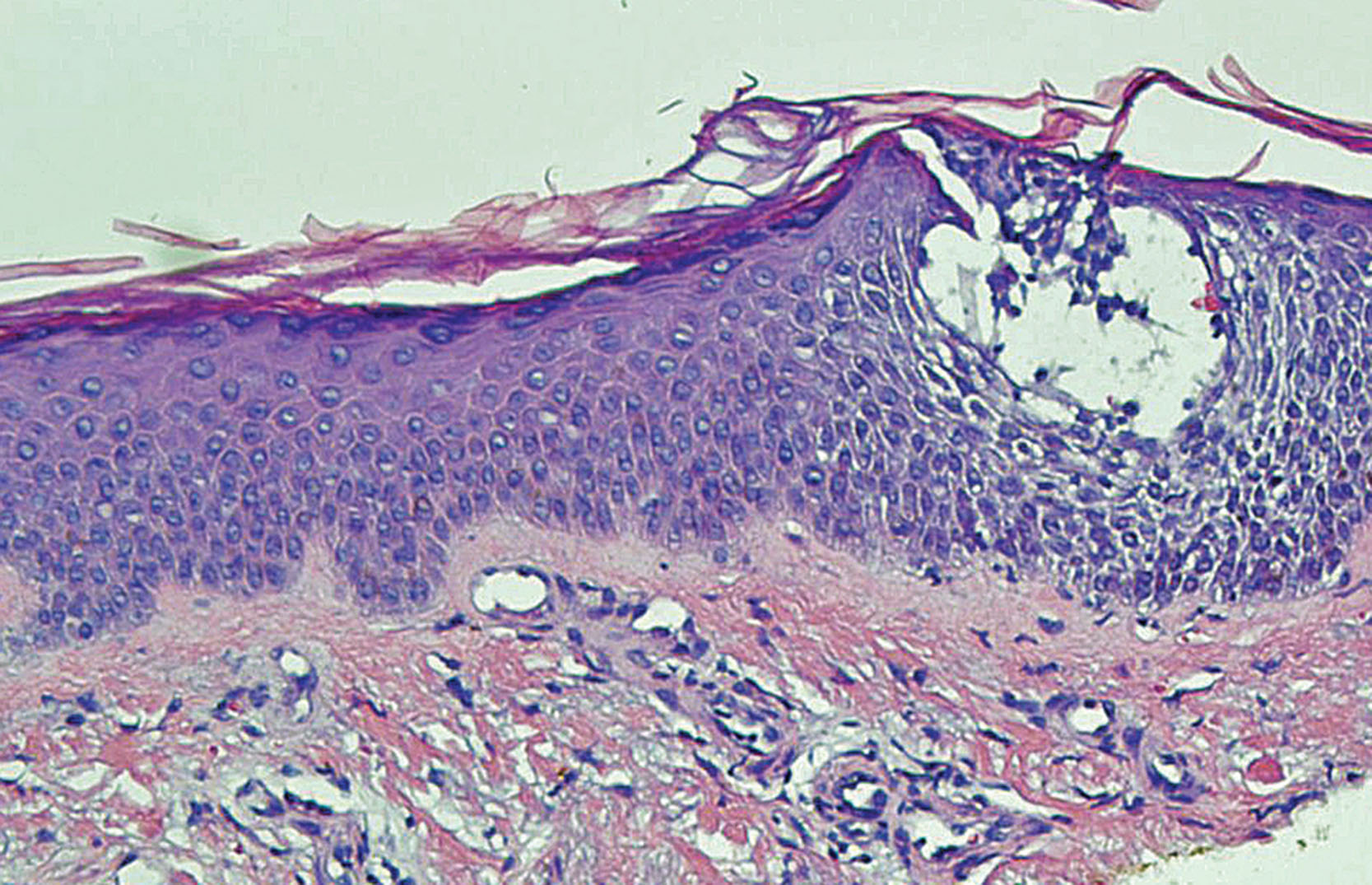
In the nonvesicular lesional skin of atopic dermatitis, one can commonly visualize a picture of inflammation with the following findings under the microscope ( Fig. 3.1 ). In the acute inflammatory lesion, the epidermis shows psoriasiform hyperplasia and intercellular edema. Epidermal and dermal white blood cell infiltrations are composed primarily of lymphocytes with occasional monocytes/macrophages, neutrophils, eosinophils, and basophils. Mast cells and Langerhans cells are present. We can also observe some vascular changes, including endothelial cell hypertrophy, rare endothelial mitosis, and large activated nuclei, suggesting a process of angiogenesis. In chronically inflamed lesion, the epidermis is visibly different from the acute lesion, with the presence of hyperkeratosis and dyskeratosis besides psoriasiform hyperplasia. Varying degrees of intercellular edema and a few lymphocytes are present. In the dermis there are a moderate number of lymphocytes and monocytes/macrophages. The number of mast cells significantly increases ( ). The thickness of the granular layer of the epidermis in atopic dermatitis lesion can be similar to that of normal individuals, unless atopic dermatitis occurs in a person who has filaggrin gene mutation with a corresponding reduction of granular cell layer ( ). More details of filaggrin mutation in atopic dermatitis are discussed in Chapter 5, Chapter 11 .
A bird’s-eye view of the atopic skin surface
Skin barrier defect and hydration problem
The initial discovery of loss-of-function genetic mutation of filaggrin in nearly 50% of white patients affected by atopic dermatitis opens a new scientific window into the disease mechanism of atopic dermatitis and atopy in general ( ). However, researchers subsequently determined that the filaggrin mutation occurred in much lower frequency in nonwhite atopic dermatitis patients ( ). Whether atopic dermatitis patients have other skin barrier protein defects aside from filaggrin is not known at the present time. Although some patients do not have a genetic mutation of skin barrier proteins, their barrier proteins can be deficient secondary to the Th2 cytokine upregulation. Experimentally, human epidermal keratinocytes, under the influence of IL4, or a combination of IL4/IL13, reduce their ability to synthesize various skin barrier proteins such as filaggrin, involucrin, and loricrin ( ). Thus, in clinically observed inflammatory skin lesions, these skin barrier proteins may in fact be reduced secondary to inflammation without the corresponding gene mutation.
Intimately related to skin barrier function, skin hydration is reduced in patients affected by atopic dermatitis. Xerosis (dry skin) is a common finding in skin of atopic dermatitis patients and is a clinical diagnostic criterion for atopic dermatitis ( ). This improper hydration condition in atopic skin, predisposed to penetration of irritants and allergens, is documented by an increase of transepidermal water loss ( ). More discussions on skin barrier defect in atopic dermatitis are available in Chapter 5, Chapter 11, Chapter 15, Chapter 22 .
Staphylococcus colonization and dysbiosis
Pathogenic Staphylococcus aureus species commonly colonize in 30% to 100% of atopic skin, and this abnormal colonization is associated with increased disease severity ( ). In addition, staphylococcal superantigen from pathogenic species can induce IL31, an itch-inducing cytokine, in skin and in peripheral blood mononuclear cells, leading to promotion of pruritus symptom ( ). The upregulation of IL31 can then induce secretion of IL1 and IL6, which could then lead to cardiovascular comorbidities (discussed later) ( ). When this invasive species takes root and expands its growth, it pushes out the skin commensal bacteria, leading to dysbiosis, a condition of alteration of resident commensal community with the disruption of symbiosis ( ). The scientific community has now recognized that the microbiota (the microorganisms of a particular site) have an important role in immune development and maturation, and that alterations of microbiota have been observed in animal models and in human patients of several immune-mediated diseases, including inflammatory bowel diseases and diabetes ( ). Since commensal bacteria serve a protective function for their hosts, their relative absence reduces one important defense against skin infection and inflammation. Together with reduction of skin barrier and skin-containing antimicrobial peptides, these deficiencies result in the clinical observation of frequent skin infection in patients affected by atopic dermatitis, including serious bacterial, fungal, and viral infections ( ). Although uncommon, systemic infections can occur and can be life threatening, and therefore should be recognized as a serious atopic dermatitis comorbidity ( ). In addition, the colonization of pathogenic S. aureus species creates substantial challenges for the diagnosis and treatment in patients of atopic dermatitis ( ). Greater details on the dysbiosis in atopic dermatitis are discussed in Chapter 6 .
A bird’s-eye view of the atopic physiology
The burdens of itch and pain
Itch and pain together generate substantial symptoms of atopic dermatitis. Recently, IL31, a cytokine upregulated in atopic dermatitis and a key component in causing pruritus, is identified as a link between activated T cells and itch in atopic skin inflammation. Pruritic symptom in atopic dermatitis links at least in part to the colonization of S. aureus , and superantigen (Staphylococcal enterotoxin B) that can induce IL31 in skin and in peripheral blood mononuclear cells ( ). In a survey of 304 patients affected by atopic dermatitis, 91% of individuals reported that itch occurred at least once daily, and greater than 50% of patients reported pain and heat sensation in association with itch symptom ( ). As we will learn in the following section, these symptoms could lead to sleep disturbance and other psychologic and physiologic health hazards. More detailed discussions on pruritus and pain in atopic dermatitis are available in Chapter 16, Chapter 25 .
Sleep disturbance and the role of circadian rhythm and melatonin
One of the major health hazards that patients with atopic dermatitis face is sleep disturbance because of the chronic nature of itchiness and other disturbing sensations such as pain and heat ( ). Reduction of sleep quality has been reported in children affected by atopic dermatitis ( ). How does sleep disturbance affect one’s circadian rhythm and overall health?
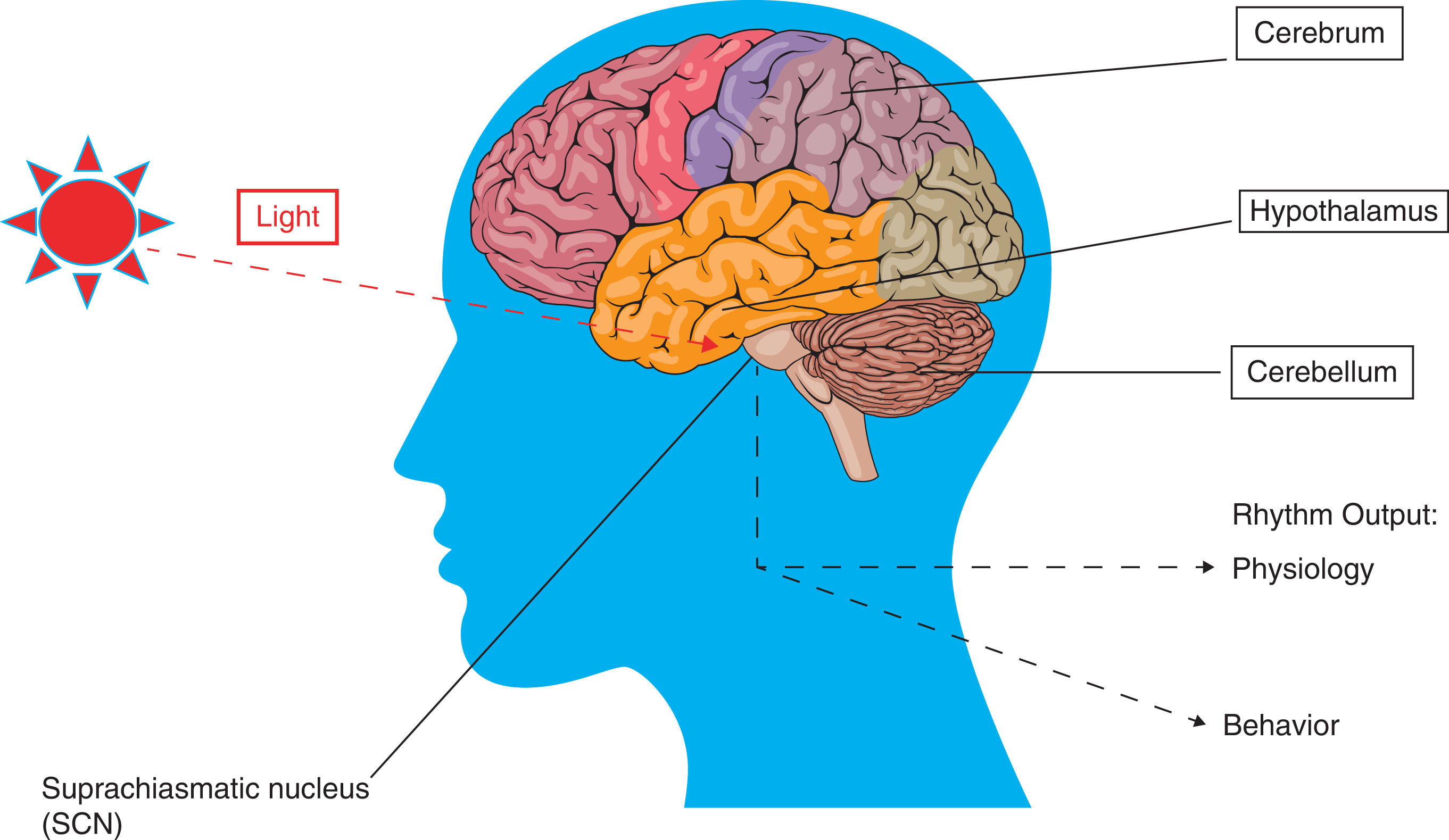
According to information provided by the National Institute of General Medical Sciences ( ), circadian rhythms are “physical, mental, and behavioral changes that follow a daily cycle.” Living organisms, plants, animals, even bacteria have followed these rhythms. A related biologic phenomenon called biologic clock is defined as an organism’s innate timing device composed of specific protein molecules that interact with different cells in the body. The relationship between these two is that the biologic clock, the internal timing machine, generates the circadian rhythm and regulates it. In living organisms, there is a so-called master clock, which in humans and vertebrate animals is formed by a group of 20,000 neurons into a structure called suprachiasmatic nucleus (SCN) in the brain’s hypothalamus. SCN receives input directly from the eyes, coordinates all the biologic clocks, and keeps the clocks in proper working order. Consequently, these rhythmic outputs affect human physiology and behavior. One example is sleep cycle. When there is less light coming from the eyes at late evening time, the stimulation of optic nerves is slower; that message is then related to SCN, which signals the brain to release more melatonin that induces drowsiness, leading to sleep ( ) ( Fig. 3.2 ).
Historically, the French astronomer and botanist Jean-Jacques de Mairan (1678–1771) was credited as being the first person to show that daily rhythms could be generated internally (i.e., endogenous rhythmicity). He observed that Mimosa pudica , a sensitive plant that folds its leaves when touched, folds up its leaves at nighttime and opens them again at daytime; however, the plant still folds its leaves at nighttime and opens them again at daytime when he placed the plant in total darkness ( ). Augustin Pyramus de Candolle (1778–1841), a Swiss botanist, later showed that by placing the sensitive plant under continuous lighting, the plant still folds its leaves at nighttime and opens them at daytime; however, this occurred with a rhythm cycle duration shorter than 24 hours, effectively demonstrating the existence of an internal rhythmic clock ( ). The first animal model that was used to illustrate the circadian rhythmicity was credited to Maynard Johnson, who published the findings in 1926 on circadian rhythm on mammals. While keeping deer mice (Peromyscus leucopus) in total darkness and constant temperature, he observed a circadian rhythm in the mice, with a cycle time slightly longer than 24 hours, further supporting an endogenously controlled rhythm ( ). Subsequently, many studies have been conducted in humans. Other key events of circadian rhythm research in the 2000s focused on SCN, molecular clock, and redox. Later, a majority of current research focused on the mechanism of biologic timing ( ). The term designated to the study of circadian rhythm is chronobiology ( ).
How does the disruption of normal sleep and circadian rhythm impact on human health? Both internal and external factors influence circadian rhythms. The principal factor for humans is daylight, which can turn on or off genes that control the molecular structure of biologic clocks, which then affects our energy, mood, emotion, and other physiologic functions. One does not need to go far to verify this: Simply recall an experience of jetlag that led to fatigue, lack of appetite, and emotional irritability. Studies conducted in the past decades have documented many diseases associated with disturbance of circadian cycle, including cardiovascular, gastrointestinal, neurologic, psychologic, neoplastic, endocrine, and immunologic diseases ( ). Atopic dermatitis, being an immune-mediated disorder, is no exception ( ). Researchers reported that during an active phase of atopic dermatitis, both melatonin and β-endorphin levels in serum reduced ( ). Studies on the circadian rhythm in atopic dermatitis patients have focused on the central rather than the peripheral circadian rhythm, and a picture of misalignment of circadian rhythm has emerged with shifts in the peaks and troughs of cortisol secretion. In addition, some atopic dermatitis patients with suppressed levels of cortisol and adrenocorticotropic hormone may have reduction of natural secretion of melatonin. Many studies utilizing the techniques of actigraphy (monitor rest/activity cycles) and polysomnography (monitor brain waves, oxygen concentration, heart and respiratory rates, and movements) in recent years have documented the deficient sleep in children affected by atopic dermatitis. Sleep efficiency is defined by the ratio of the actual sleep time to the total record time in bed by polysomnography. In children, sleep disturbance because of atopic dermatitis has been associated with behavior problems, neurocognitive defect, lower IQ, attention-deficit hyperactivity, poorer performance of schoolwork, obesity, hypertension, and impaired linear growth ( ).
The cardiovascular and cerebrovascular comorbidities
As an inflammatory skin disease, atopic dermatitis is characterized by increased serum levels of cytokines such as IL1 and IL6 ( ). It is therefore logical for one to speculate that these proinflammatory and proatherogenic cytokines (IL1 and IL6) could lead to increased cardiovascular and/or cerebrovascular comorbidity, as supported by findings of an in vitro study ( ) and by evidence that such cytokine upregulation in another inflammatory skin disease (psoriasis) also linked to cardiovascular comorbidity ( ). Other evidence supporting this link is that staphylococcal superantigen derived from S. aureus can induce IL31 from human monocytes/macrophages. Consequently, these induced IL31 cytokines can then enhance IL1 and IL6 secretions that might contribute to these vascular comorbidities ( ). Recent clinical studies seem to point to evidence to support this link ( ). Keep in mind, however, some studies reported no increase or minimum increase of cardiovascular risk in atopic dermatitis patients ( ), and some academicians have questioned the adequacy of current epidemiologic tools utilized in the risk analysis ( ). Nevertheless, the majority of evidence seems to settle in that the more severe the atopic dermatitis, the higher the risk of cardiovascular comorbidity ( ). This conclusion, similar to the reports from psoriasis studies ( ), seems to make sense from the immune mechanism perspective. The more severe disease would allow more proinflammatory and proatherogenic cytokines to be produced. Once these cytokines reach the blood vessels they could lead to cardiovascular or cerebrovascular comorbidities.
A bird’s-eye view of the atopic immune deviation
The hygiene hypothesis
The hygiene hypothesis of allergy, first proposed by , speculates that the arising of atopic dermatitis is due in major part to the modern family practice of keeping children in an excessively clean (hygienic) condition, so much so that the children grow up lacking exposure to certain pathogens ( ). The absence of such common pathogens challenges children’s immune systems, as the theory speculated, and can then lead to deviation from a balanced milieu toward to a Th2-dominant immune milieu. This theory is based, in part, on the epidemiologic findings that the substantial increase of atopic dermatitis prevalence is primarily observed in developed nations (which tend to have more stringent hygiene practices than developing nations) and that migrants moving from a low-incidence country to a high-incidence country tend to acquire the disease ( ). One survey pointed out an inverse relationship between development of atopic dermatitis and exposures of endotoxin, early day care, farm animal and dog in early age, and helminth infections ( ).
Innate immune deviation
The innate immune system is the first line of immune defense against invading pathogens, and it does not require prior training or memory to act effectively. Antimicrobial peptides are one of the major components that have a role in the skin immune defense ( ). The altered immune milieu, particularly an increase of IL10 in atopic dermatitis, can suppress the expression of these antimicrobial peptides ( ). Studies pointed to a deficiency of human skin-producing cathelicidin (LL37) and human β-defensin-2 in patients of atopic dermatitis, particularly on lesional skin ( ). Such deficiencies have been linked to the serious bacterial, fungal, and viral infections ( ).
Adaptive immune deviation
Adaptive immune system requires a prior encounter of pathogens to develop a long-term cellular memory, such that a subsequent challenge by the same pathogens will lead to rapid generation and movement of immune armies, lymphocytes or antibodies, aiming to eliminate the invading enemies. Once the invading microorganisms are eliminated, the immune system will naturally shut down the inflammatory process. In chronic inflammatory disease atopic dermatitis, however, such an inflammatory process, once activated, will remain activated. Commonly, T-cell activation, IgE, Th2, Th17, and Th22 were upregulated, with predominant increase of IL4, IL5, IL6, IL10, IL13, and IL31 and variable increase in IL17 and IL22 ( ; ; ; ; ). The continuous presence of these cytokines thus fuels the ongoing inflammation until external antiinflammatory medications stop it.
The early reports of total serum IgE level showed a substantial upregulation in patients with atopic dermatitis, and on the average the atopic dermatitis patients have 6793 ng/mL and 493 IU mL, compared to normal average levels of 160 ng/mL and 122 IU mL, respectively ( ). More discussions of immune factors in atopic dermatitis are available in Chapter 13, Chapter 14 .
The skin-gut-lung link in atopic march
Literature accumulated over years of studies has pointed to a link between the skin, the intestine, and the lung in terms of the immune deviation and the resulting atopy manifestations, and it has been postulated that initial disruption of the skin epidermal barrier permits allergen sensitization and colonization by pathogens. As hypothesized, this initial skin disruption induces a Th2 inflammatory response and a thymic stromal lymphopoietin-mediated pathway that further promotes barrier breakdown at distant sites, including the intestinal and respiratory tract ( ). This may explain the atopic march phenomenon, defined as the initial clinical manifestation of skin atopy (i.e., atopic dermatitis) that will later expand to affect other organ systems, including the gastrointestinal and respiratory systems, resulting in food allergy, rhinitis, and asthma, as observed by clinicians ( ). Additional discussions on the skin-gut-lung link are provided in Chapter 15 .
Summary
In summary, atopic dermatitis is a chronic, itchy, inflammatory skin disease characterized by skin barrier defect, hydration deficiency, pathogenic bacterial colonization, chronic pruritus and skin inflammation, inefficient innate immune functions, and adaptive immune deviation. In addition, atopic dermatitis carries a heavy disease burden to the patient, as the itch and pain cause sleep disturbance and circadian rhythm alterations as well as cardiovascular and cerebrovascular comorbidities. Atopic dermatitis is not only a skin disease; it also has substantial systemic implications, as it can expand to affect the gastrointestinal and respiratory systems in the development of food allergy, rhinitis, and asthma. As detailed in Chapter 2 , accumulative evidence gathered from clinical and laboratory investigations has pointed to a need to redefine this entity or to determine subsets within this big umbrella of atopic dermatitis. Overall, however, the general characteristics of atopic dermatitis described in this chapter, to a large extent, still hold true.
Further readings
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


