Abstract
Cutaneous xanthomas develop as a result of intracellular and dermal deposition of lipid. One of their major distinguishing clinical features is a characteristic yellow to orange hue. They are a reflection of a range of disorders that affect lipid metabolism, from primary causes such as familial hypercholesterolemia to secondary etiologies such as cholestasis or medications (e.g. retinoids). The major forms of xanthomas associated with hyperlipidemia are: eruptive, tuberous, tendinous, and plane (including xanthelasma). In addition, normolipemic plane xanthoma can develop in association with monoclonal gammopathies. Histologically, lipid-laden macrophages (foam cells) are seen in the dermis. The therapeutic approach is dependent upon the underlying cause and includes medications (e.g. HMG-CoA reductase inhibitors, insulin), dietary restrictions, and in the case of xanthelasma, chemical or physical destruction.
Keywords
xanthomas, hypercholesterolemia, hyperlipidemia, hypertriglyceridemia, eruptive xanthoma, tuberous xanthoma, tuberoeruptive xanthoma, tendinous xanthoma, plane xanthoma, xanthelasma, normolipemic plane xanthoma, verruciform xanthoma
- ▪
Cutaneous xanthomas can signal the presence of an underlying hyperlipidemia or monoclonal gammopathy
- ▪
An understanding of basic lipid metabolism provides insight into the underlying hyperlipoproteinemias as well as the formation of xanthomas
- ▪
The major forms of xanthomas associated with hyperlipidemia are: eruptive, tuberous, tendinous and plane (including xanthelasma)
- ▪
Normolipemic plane xanthomas occur in association with monoclonal gammopathies
- ▪
Histologically, lipid-laden macrophages (foam cells) are seen in the dermis
- ▪
Prompt recognition and proper treatment can lead to xanthoma resolution as well as prevention of potentially life-threatening complications
Introduction
Cutaneous xanthomas develop as a result of deposition of lipid in the dermis, primarily within macrophages (foam cells) but also extracellularly. One of their major distinguishing clinical features is a characteristic yellow to orange hue. Xanthomas may present with a variety of morphologies, from macules and papules to plaques and nodules. As discussed below, the morphology and anatomic location of the lesions often suggest the type of underlying lipid disorder or the presence of a paraproteinemia.
Xanthomas can develop in the setting of primary or secondary disorders of lipid metabolism. Thus, early recognition of these lesions can make a significant impact on the diagnosis, management, and prognosis of patients who suffer from an underlying disease. It is therefore important for dermatologists to become familiar with the basic concepts of lipid metabolism and the associated disease states, as well as to be able to recognize the often pathognomonic cutaneous findings.
Epidemiology
Hyperlipidemia is quite common in the general population. In North America alone, it is estimated that over 100 million people currently have an elevated serum cholesterol level >200 mg/dl. Despite the large number of people who suffer from hyperlipidemia, only a minority will develop cutaneous xanthomas. Also, because the exact mechanism by which xanthomas form is not yet fully understood, it is not always possible to predict who will develop them. Xanthomas are thought to result from the permeation of circulating plasma lipoproteins through dermal capillary blood vessels followed by phagocytosis of the lipoproteins by macrophages, forming lipid-laden cells known as foam cells . However, the precise steps and their regulation are still an area of investigation.
Pathogenesis
There is strong evidence to support the theory that the lipids found in the various xanthomas are the same as those in the circulation . The majority of plasma lipids are transported in complex structures known as lipoproteins. The basic structure of the lipoprotein allows the delivery of triglycerides and cholesterol to peripheral cells for their metabolic needs. This structure consists of a hydrophilic outer shell and a hydrophobic core. The outer shell consists of phospholipids, free cholesterol, and non-covalently linked specialized proteins known as apolipoproteins or apoproteins (apo). The inner core contains triglycerides and cholesterol esters.
Lipoproteins differ in their core lipid content. Triglycerides are the major core lipids in chylomicrons and very-low-density lipoproteins (VLDLs), while cholesterol esters dominate the core of low-density lipoproteins (LDLs), high-density lipoproteins (HDLs), and remnants of chylomicrons and VLDLs. The apoproteins found in the outer shell can also differ amongst the various lipoproteins ( Table 92.1 ). These apoproteins serve several important functions, such as mediating the binding of lipoproteins to their respective receptors in target organs and activating enzymes involved in their metabolism.
| IMPORTANT APOPROTEINS | ||
|---|---|---|
| Apoprotein | Lipoprotein association | Function and comments |
| A-I | Chylomicrons, HDLs | Major protein of HDL; activates l ecithin: c holesterol a cyl t ransferase (LCAT) |
| B-48 | Chylomicrons, chylomicron remnants | Unique marker for chylomicrons |
| B-100 | VLDLs, IDLs, and LDLs | Major protein of LDL; binds to LDL receptor |
| C-II | Chylomicrons, VLDLs, IDLs, and HDLs | Activates lipoprotein lipase |
| E (at least 3 alleles [E 2 , E 3 , E 4 ]) | Chylomicrons, chylomicron remnants, VLDLs, IDLs, and HDLs | Binds to LDL receptor |
There are two major pathways of lipoprotein synthesis ( Fig. 92.1A ). The exogenous pathway begins with dietary fat intake. Through the action of pancreatic lipase and bile acids, dietary triglycerides are degraded to fatty acids and monoglycerides. After absorption by the intestinal epithelium, the triglycerides are reformed and packaged with a small amount of cholesterol esters into the central core of a chylomicron. The outer shell of the chylomicron consists of phospholipids, free cholesterol, and several apoproteins, including B-48, E, A-I, A-II, and C-II.
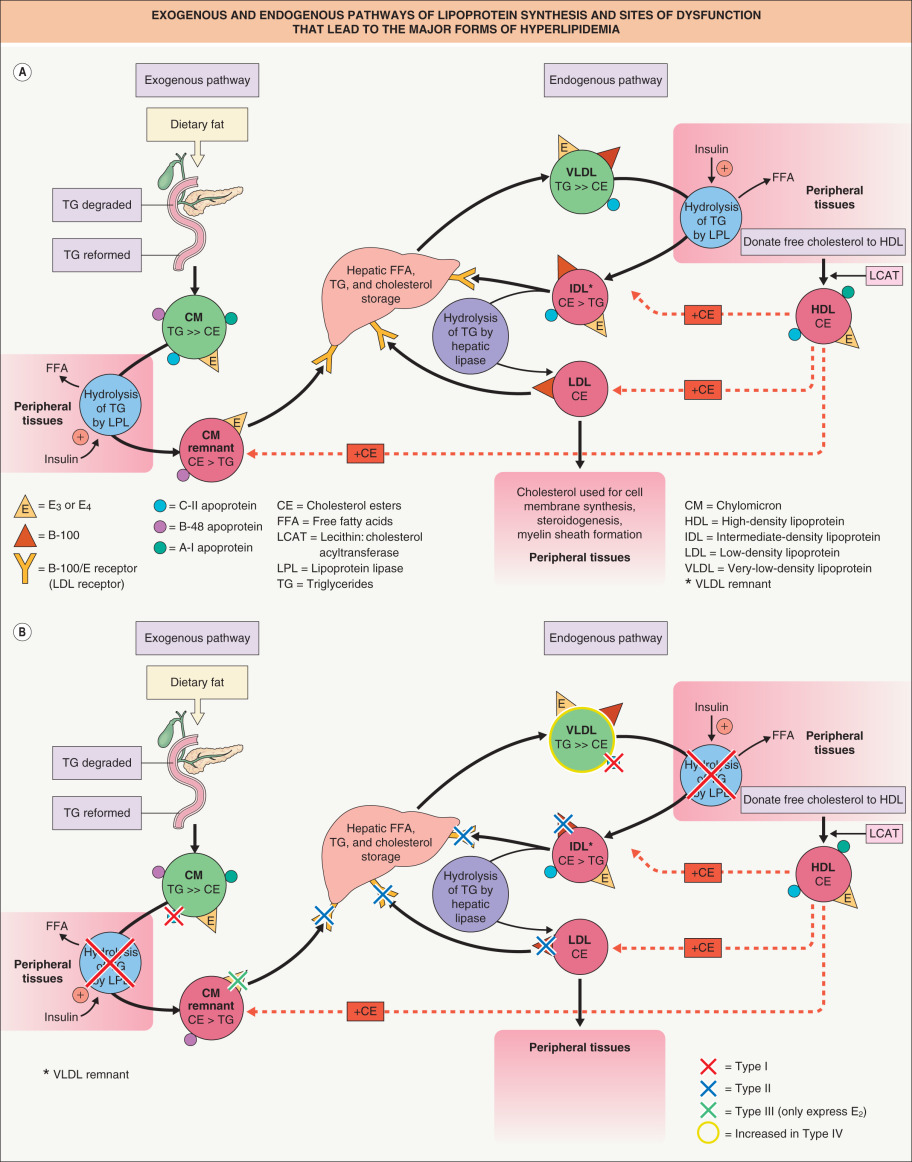
Chylomicrons then enter the lymphatics and eventually the systemic circulation via the thoracic duct. Once in the circulation, hydrolysis of the core triglycerides occurs, releasing free fatty acids to the peripheral tissues. This is mediated through the action of the enzyme lipoprotein lipase that is bound to capillary endothelium. The activation of the lipoprotein lipase system is complex and involves not only hormones such as insulin, but also apoproteins such as C-II, located on the lipoprotein outer surface, and GPIHBP1, a protein expressed on endothelial cells that binds lipoprotein lipase and shuttles it to its site of action in the capillary lumen.
After hydrolysis of approximately 70% of the original triglyceride content, a chylomicron “remnant” exists. The central core now contains predominantly cholesterol ester that has been acquired from circulating HDL molecules. The chylomicron remnant is taken up by the liver via specialized high-affinity apo B-100/E receptors that recognize the apoproteins E 3 or E 4 on the remnant’s outer shell. Once in the liver, the remaining lipids enter hepatic storage and apoproteins such as B-48 are degraded.
The endogenous pathway begins with the hepatic formation of VLDL particles. The central core of the VLDL consists primarily of triglycerides, which are derived from circulating free fatty acids and hepatic triglyceride stores. Important apoproteins found on the outer shell include B-100, E, and C-II. In a fashion similar to the chylomicron, lipoprotein lipase mediates hydrolysis of the VLDL molecule, removing the majority of its triglyceride content, and its cholesterol esters are acquired from HDL molecules. Lipoprotein lipase activation requires the presence of apo C-II on the VLDL outer shell. After removal of the majority of the triglyceride content, the VLDL “remnant”, also known as an intermediate-density lipoprotein (IDL), can then be taken up by the liver via apo B-100/E receptors and degraded. IDLs that escape uptake by the hepatocyte are stripped of their remaining core triglycerides by extracellular hepatic lipases and enter the circulation as LDLs.
The LDL contains predominantly cholesterol ester in its central core and expresses B-100 on its surface. LDL delivers cholesterol ester to peripheral tissues, where it can be converted to free cholesterol. Cholesterol has several important functions within the body, including being an essential component of cell membrane bilayers. It is also important in the production of the myelin sheath of nerves, adrenal and gonadal steroidogenesis, and the production of bile acids. Hepatocytes play the major role in the catabolism of LDLs. Their uptake is mediated through the high-affinity apo B-100/E receptor found on the cell surface of the hepatocytes. Free cholesterol in excess of metabolic needs is re-esterified for storage.
HDLs serve several important functions in cholesterol metabolism. One of the primary functions of HDLs is the removal of cholesterol from the peripheral tissues. During this process, free cholesterol and phospholipids are transferred from the cell membranes of peripheral cells to the HDL molecules. The free cholesterol is then esterified by the enzyme l ecithin: c holesterol a cyl t ransferase, or LCAT. This enzyme requires the presence of the HDL apoprotein A-I. HDL molecules then transfer the cholesterol esters to other lipoproteins such as LDLs and remnants of chylomicrons or VLDLs for transportation back to the liver.
The liver plays the central role in the overall cholesterol economy. Hepatic intracellular cholesterol levels have a direct impact on the activity of HMG-CoA reductase, the rate-limiting enzyme of cholesterol synthesis, and on the expression of the high-affinity apo B-100/E receptor. When intracellular cholesterol levels are low, HMG-CoA reductase becomes activated and high-affinity apo B-100/E receptor expression increases. The increase in high-affinity receptors leads to increased uptake of cholesterol-containing lipoproteins such as chylomicron remnants, IDLs and LDLs. This is followed by the lowering of plasma cholesterol levels. As discussed later, this mechanism will be the basis for many of the pharmacologic interventions aimed at lowering cholesterol levels.
Clinical Features
Due to the complexity of cholesterol homeostasis, there are several possible ways in which hyperlipidemia may occur, from inherited disorders to metabolic diseases such as diabetes mellitus. Genetic mutations can affect important enzymes, receptors or receptor ligands, with such defects leading to the overproduction of lipoproteins or the inhibition of their clearance ( Fig. 92.1B ). Each possible defect would lead to a different abnormal lipid profile.
In 1965, Lees and Frederickson published a system for classifying various disorders of lipid metabolism based upon the electrophoretic migration of the serum lipoproteins present. This system for phenotyping hyperlipoproteinemias is used today in a modified form ( Table 92.2 ). In the next section, descriptions of underlying lipid disorders will reference not only the Frederickson classification system, but also the specific molecular defects when possible.
| IMPORTANT HYPERLIPOPROTEINEMIAS | ||||
|---|---|---|---|---|
| Type | Pathogenesis | Laboratory findings | Clinical findings | |
| Skin (types of xanthoma) | Systemic | |||
| Type I (familial LPL deficiency, familial hyperchylomicronemia) |
| Slow chylomicron clearance Reduced LDL and HDL levels Hypertriglyceridemia | Eruptive | No increased risk of coronary artery disease Recurrent pancreatitis |
| Type II (familial hypercholesterolemia) | Reduced LDL clearance Hypercholesterolemia | Tendinous, tuberoeruptive, tuberous, plane (xanthelasma, intertriginous areas, interdigital web spaces † ) | Atherosclerosis of peripheral and coronary arteries | |
| Type III (familial dysbetalipoproteinemia, remnant removal disease, broad beta disease, apo E deficiency) | Hepatic remnant clearance impaired due to apo E abnormality; vast majority of patients only express the apo E 2 isoform that interacts poorly with the apo E receptor (AR>>AD) | Elevated levels of chylomicron remnants and IDLs Hypercholesterolemia Hypertriglyceridemia | Tuberoeruptive, tuberous, plane (palmar creases) – most characteristic Tendinous | Atherosclerosis of peripheral and coronary arteries |
| Type IV (endogenous familial hypertriglyceridemia) | Elevated production of VLDL associated with glucose intolerance and hyperinsulinemia; may be associated with heterozygous variants, e.g. in apo A-V, LPL, or the glucokinase regulator protein | Increased VLDLs Hypertriglyceridemia | Eruptive | Frequently associated with type 2 non-insulin-dependent diabetes mellitus, obesity, alcoholism (see Fig. 92.4 ) |
| Type V | Elevated chylomicrons and VLDLs; may be associated with heterozygous variants, e.g. in apo A-V, LPL, or the glucokinase regulator protein | Decreased LDLs and HDLs Hypertriglyceridemia | Eruptive | Diabetes mellitus |
^ In patients with the LDLR IVS14+1G-A mutation, the phenotype can be altered by SNPs in the genes that encode apo A-II, cytoplasmic epoxide hydrolase 2, or growth hormone receptor.
* Gain-of-function mutations cause autosomal dominant hypercholesterolemia , whereas loss-of-function mutations (most prevalent in African-Americans) result in low LDL levels .
With the exception of the homozygous form of familial hypercholesterolemia (type II), most cutaneous xanthomas do not appear until adulthood. Once the diagnosis is established, treatment of associated disorders (e.g. metabolic syndrome [see Table 53.5 ]) should reduce the incidence of potential systemic sequelae such as myocardial infarctions, cerebrovascular accidents, and hepatic steatosis.
Eruptive Xanthomas
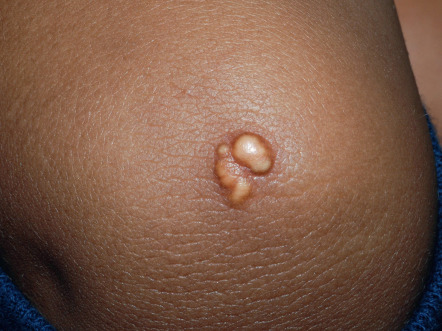
Eruptive xanthomas appear as erythematous to yellow papules, approximately 1 to 5 mm in diameter ( Figs 92.2 & 92.3 ). They are usually distributed on the extensor surfaces of the extremities, buttocks, and hands. Early in their development, lesions may have an inflammatory halo (likely due to their triglyceride component), which may be accompanied by tenderness and pruritus. The Koebner phenomenon has been reported to occur with eruptive xanthomas .
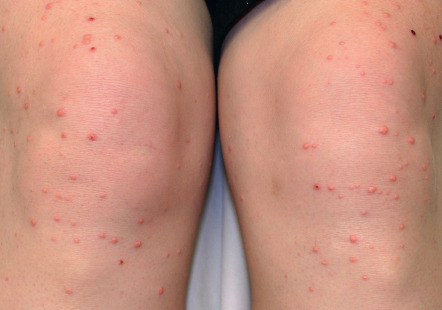
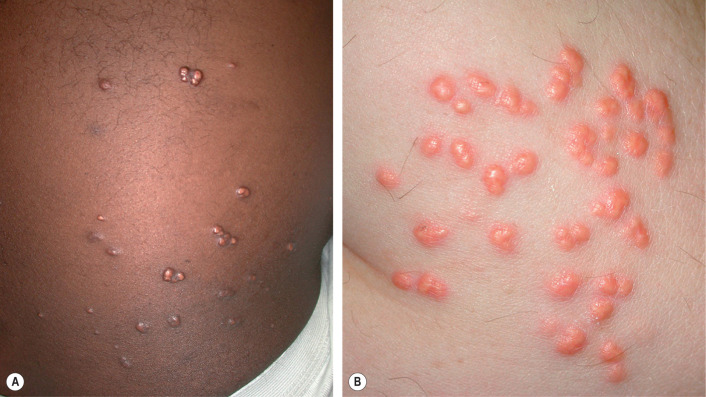
Eruptive xanthomas can be seen in the setting of primary or secondary hypertriglyceridemia . Triglyceride levels in patients with eruptive xanthomas often exceed 3000 to 4000 mg/dl. In the Frederickson classification of hyperlipidemias, hypertriglyceridemia can be seen in type I (elevated chylomicrons), type IV (elevated VLDLs) and type V (elevated chylomicrons and VLDLs).
One reason for elevated triglyceride levels is failure to remove such lipids from the circulation ( Fig. 92.4 ). Deficient activity of lipoprotein lipase will lead to accumulation of triglyceride-rich chylomicrons and VLDLs (see Fig. 92.1B ). This can be related to either abnormalities in the enzyme itself, as in lipoprotein lipase deficiency ( chylomicronemia syndrome ), or in other controlling factors such as dysfunctional apoprotein C-II or impaired insulin activity . In addition to deficiency of GPIHBP1 , autoantibodies against GPIHBP1 can also lead to hypertriglyceridemia .
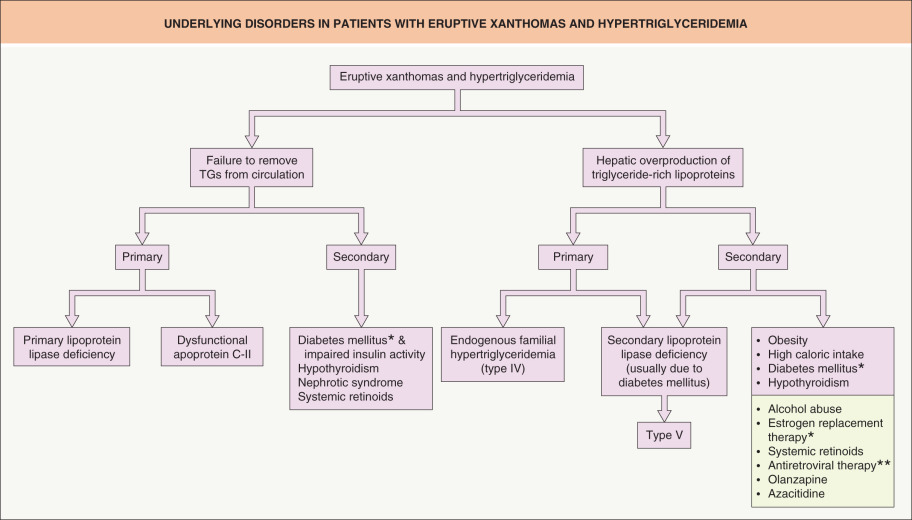
Another reason for increased triglyceride levels is hepatic overproduction of triglyceride-rich lipoproteins via the endogenous pathway. In endogenous familial hypertriglyceridemia , a genetic defect exists that causes the liver to respond abnormally to dietary carbohydrates and insulin, with overproduction of hepatic VLDLs. The result is a Frederickson type IV pattern of hypertriglyceridemia. Secondary acquired defects in lipoprotein lipase activity, such as those due to diabetes mellitus, are not uncommon in these patients. With this second insult, the lipoprotein lipase system can become saturated and, as a result, no longer handles dietary lipids, leading to chylomicron elevations as well. This pattern is classified as a Frederickson type V phenotype.
Environmental factors and underlying diseases commonly exacerbate genetic defects of triglyceride metabolism, leading to worsening of the hypertriglyceridemia with eruptive xanthoma formation (see Fig. 92.4 ). Such factors include obesity, high caloric intake, diabetes mellitus, alcohol abuse, oral estrogen replacement, and systemic medications that can lead to hypertriglyceridemia (e.g. retinoids, protease inhibitors, olanzapine). The resultant pattern usually leads to a Frederickson type IV phenotype. Oral retinoid therapy, especially bexarotene, can elevate triglyceride levels through an elevation in hepatic VLDL secretion. With isotretinoin, this elevation seems to be more prevalent in genetically predisposed individuals and may signal an increased risk for future metabolic syndrome . Two of the five criteria for the clinical diagnosis of metabolic syndrome are lipid abnormalities – elevated triglycerides and reduced HDLs (see Table 53.5 ).
The treatment of eruptive xanthomas involves the identification and treatment of the underlying causes of the hypertriglyceridemia (see Fig. 92.4 ). Failure to recognize and treat the patient with hypertriglyceridemia could lead to complications such as acute pancreatitis. Pharmacologic and dietary lowering of the circulating triglycerides to reasonable levels will result in the prompt resolution of the eruptive lesions.
Tuberous/Tuberoeruptive Xanthomas
Tuberoeruptive and tuberous xanthomas are clinically and pathologically related and often described as being on a continuum. Tuberoeruptive xanthomas present as pink–yellow papules or nodules on extensor surfaces, especially the elbows and knees ( Fig. 92.5 ). Tuberous lesions are noted to be larger than tuberoeruptive lesions and may exceed 3 cm in diameter ( Fig. 92.6 ). Together, these lesions can be seen in hypercholesterolemic states such as dysbetalipoproteinemia (Frederickson type III) and familial hypercholesterolemia (Frederickson type II; see below). In contrast to eruptive xanthomas, tuberous xanthomas are usually slow to regress following institution of appropriate therapy.