Abstract
Leukoderma and hypopigmentation refer to lightening of the skin, which is classically due to decreased epidermal melanin (hypomelanosis) but may also result from decreased cutaneous blood supply. Melanocytopenic hypomelanosis is characterized by reduced or absent vs a reduction in the number of melanocytes, whereas melanopenic hypomelanosis features a normal number of melanocytes but decreased melanin synthesis or transfer to keratinocytes. The age of onset (congenital or acquired), degree of pigment loss (hypo- versus depigmentation), presence or absence of preceding inflammation, anatomic location, and distribution pattern – circumscribed, diffuse, linear, or guttate – represent key considerations in the evaluation of a patient with hypomelanosis. This chapter first discusses vitiligo, a common multifactorial disorder involving the autoimmune destruction of melanocytes that presents with depigmented macules and patches in a localized or generalized distribution. Hereditary, nutritional, postinflammatory, infectious, and chemical forms of hypomelanosis as well as halo nevi and melanoma-associated leukoderma are also reviewed.
Keywords
hypopigmentation, leukoderma, hypomelanosis, depigmentation, vitiligo, oculocutaneous albinism, piebaldism, Waardenburg syndrome, Hermansky–Pudlak syndrome, Griscelli syndrome, Chédiak–Higashi syndrome, ash leaf spot, nevus depigmentosus, linear nevoid hypopigmentation, hypomelanosis of Ito, segmental pigmentation disorder, progressive macular hypomelanosis, postinflammatory hypomelanosis, pityriasis alba, melanoma-associated leukoderma, halo nevus, idiopathic guttate hypomelanosis
- ▪
Leukoderma and hypopigmentation refer to lightening of the skin, which is classically due to decreased epidermal melanin (hypomelanosis)
- ▪
Melanocytopenic hypomelanosis, characterized by a reduction in the number of melanocytes, can result from a variety of defects in pigment cell differentiation, proliferation, migration, and/or survival
- ▪
Melanopenic hypomelanosis features a normal number of melanocytes but decreased melanin synthesis or transfer to keratinocytes, and the underlying abnormalities often involve melanosomes
- ▪
The age of onset (congenital or acquired), degree of pigment loss (hypo- versus amelanosis), presence or absence of preceding inflammation, anatomic location, and distribution pattern – circumscribed, diffuse, linear, or guttate – represent key considerations in the evaluation of a patient with hypopigmentation
- ▪
Vitiligo is a common multifactorial disorder involving the autoimmune destruction of melanocytes; patients present with circumscribed depigmented macules and patches in a localized (including segmental) or generalized distribution
- ▪
Presentations of hereditary hypomelanosis range from congenital, stable, circumscribed areas of amelanosis in piebaldism and Waardenburg syndrome to diffuse pigmentary dilution of the skin, hair, and/or eyes in the various forms of oculocutaneous albinism and related conditions
- ▪
Postinflammatory, infectious, and chemical hypomelanosis as well as halo nevi and melanoma-associated leukoderma represent other etiologies of circumscribed hypo- or amelanosis
- ▪
Linear nevoid hypopigmentation and idiopathic guttate hypomelanosis are the most common causes of linear and guttate hypopigmentation
Introduction
Leukoderma and hypopigmentation are general terms used to designate disorders characterized by lightening of the skin. They are classically the result of decreased epidermal melanin content (melanin-related), but they may be secondary to a decreased blood supply to the skin (hemoglobin-related). Hypomelanosis is a more specific term that denotes a reduction of melanin within the skin; amelanosis signifies the total absence of melanin. Depigmentation usually implies a total loss of skin color, most commonly due to disappearance of pre-existing melanin pigmentation, as in vitiligo. The term pigmentary dilution is used to describe a generalized lightening of the skin and hair, as in oculocutaneous albinism; this may only be apparent if the affected individuals are compared with unaffected relatives.
Cutaneous hypomelanosis is often classified into two groups:
- •
melanocytopenic hypomelanosis , caused by a reduction in the number of epidermal and/or follicular melanocytes
- •
melanopenic hypomelanosis , in which the number of epidermal and/or follicular melanocytes is normal, but the pigment cells fail to synthesize normal amounts of melanin and/or transfer it to surrounding keratinocytes.
Diagnosis of Leukodermas
Any patient with a leukoderma should be fully examined under visible light and using a Wood’s lamp (~365 nm). The latter is particularly useful in circumscribed leukodermas, individuals who have very lightly pigmented skin (phototypes I or II), and neonates. Under visible light, it is sometimes difficult to distinguish between hypomelanosis and amelanosis, but the greater the loss of epidermal pigmentation, the more marked the contrast on Wood’s lamp examination. This technique is also helpful in differentiating hypomelanotic macules from hemoglobin-related leukodermas; for example, nevus anemicus becomes inapparent.
Most leukodermas are diagnosed clinically following a complete history and physical examination. Determining whether the distribution pattern is circumscribed (e.g. vitiligo), diffuse (e.g. albinism), linear , or guttate (e.g. idiopathic guttate hypomelanosis) helps to narrow the differential diagnosis. The age of onset, presence or absence of preceding inflammation, anatomic location, and degree of pigment loss represent other pertinent features. Histologic examination of involved skin is most useful for several of the hypomelanoses associated with inflammatory processes (e.g. sarcoidosis, lichen sclerosus, mycosis fungoides).
Vitiligo
Vitiligo is an acquired disorder characterized by circumscribed depigmented macules and patches that result from the loss of functional melanocytes.
Epidemiology
Worldwide vitiligo affects approximately 0.5–2% of the general population , and it may appear any time from shortly after birth to late adulthood. The average age of onset is ~20 years. Although patients with vitiligo may attribute the onset of their disease to specific life events (e.g. physical injury, sunburn, emotional distress, illness, pregnancy), with the exception of the Koebner phenomenon, there is no proof that these factors cause or precipitate vitiligo.
Pathogenesis
Vitiligo is a multifactorial disorder related to both genetic and nongenetic factors. It is generally agreed that there is an absence of functional melanocytes in vitiligo skin and that the loss of histochemically recognizable melanocytes is the result of their destruction.
Genetics of vitiligo
Both twin and family studies point to the importance of genetic factors in the development of vitiligo . For example, a survey in the US and UK found that 7% of the first-degree relatives of vitiligo probands had vitiligo. However, a 23% concordance rate in monozygotic twins supported the additional role of environmental factors . Genome-wide linkage analyses have been performed in multiple patient populations, resulting in the identification of a number of susceptibility loci and candidate genes. Many of these genes are involved in melanogenesis, immune regulation, or apoptosis and have been associated with other pigmentary, autoimmune, or autoinflammatory disorders ( Table 66.1 ).
| SELECTED SUSCEPTIBILITY LOCI AND CANDIDATE GENES FOR VITILIGO | ||
| Chromosomal location | Candidate gene | Defective protein |
| Involved in immune regulation | ||
| 1p13 | PTPN22 | Protein tyrosine phosphatase, non-receptor type 22 |
| 1p31.3 | FOXD3 * | Forkhead box D3 * |
| 2q24 | IFIH1 | Interferon induced with helicase C domain 1 |
| 2q33.2 | CTLA4 | Cytotoxic T-lymphocyte-associated protein 4 |
| 3p14.1 | FOXP1 | Forkhead box P1 |
| 3q13.33 | CD80 | CD80 molecule |
| 3q27–q28 | LPP | LIM domain containing preferred translocation partner in lipoma |
| 4p16.1 | CLNK | Cytokine dependent hematopoietic cell linker |
| 5q22.1 | TSLP | Thymic stromal lymphopoietin |
| 6q15 | BACH2 | BTB domain and CNC homolog 2 |
| 6p21.3 | HLA (various genes), BTNL2 | Major histocompatibility complex classes I–III, butyrophilin-like 2 |
| 6q27 | CCR6 | Chemokine (C-C motif) receptor 6 |
| SMOC2 | SPARC related modular calcium binding 2 | |
| 8q24 | SLA | Src-like-adaptor |
| 10p15–p14 | IL2RA | Interleukin 2 receptor α |
| 11p13 | CD44 | CD44 molecule |
| 11q23.3 | CXCR5 | C-X-C motif chemokine receptor 5 |
| 12q13 | IKZF4 | IKAROS family zinc finger 4 |
| 12q24 | SH2B3 | SH2B adaptor protein 3 |
| 17p13 | NLRP1 ( formerly NALP1) | NOD-like receptor family, pyrin domain containing 1 |
| 18q21.33 | TNFRSF11A | TNF receptor superfamily member 11a (RANK) |
| 19p13.3 | TICAM1 | Toll-like receptor adaptor molecule 1 |
| 20q13.13 | PTPN1 | Protein tyrosine phosphatase, non-receptor type 1 |
| 21q22.3 | UBASH3A, AIRE | Ubiquitin associated and SH3 domain containing A, autoimmune regulator |
| 22q12.1 | XBP1 | X-box binding protein 1 |
| 22q13.1 | C1QTNF6 | C1q and tumor necrosis factor related protein 6 |
| 22q13.2 | TOB2 | Transducer of ERBB2, 2 |
| Xp11.23 | FOXP3 | Forkhead box P3 |
| Involved in apoptosis and/or cytotoxicity | ||
| 1q24.3 | FASL | Fas ligand |
| 1p36.23 | RERE | Arginine–glutamic acid dipeptide (RE) repeats |
| 10q22.1 | SLC29A3 | Solute carrier family 29 member 3 |
| 10q25 | CASP7 | Caspase 7 |
| 14q11.2 | GZMB | Granzyme B |
| Melanocyte related | ||
| 6p25.3 | IRF4 | Interferon regulatory factor 4 |
| 6q27 | FGFR1OP, RNASET2 | Fibroblast growth factor receptor 1 oncogene partner, ribonuclease T2 |
| 10q22.3 | ZMIZ1 | Zinc finger MIZ-type containing 1 |
| 11q14–q21 | TYR | Tyrosinase |
| 12q13.2 | PMEL | Premelanosome protein |
| 15q13.1 | OCA2, HERC2 | Oculocutaneous albinism 2 transmembrane protein, HECT and RLD domain containing E3 ubiquitin protein ligase 2 |
| 16q24.3 | MC1R | Melanocortin-1 receptor |
| 20q11.22 | ASIP | Agouti signaling protein |
* In one family to date, a functional variant in the promoter of the FOXD3 melanoblast developmental regulator gene was associated with autosomal dominant inheritance of an atypical vitiligo phenotype (early onset, widespread, progressive depigmentation).
Pathogenic hypotheses for vitiligo
Many pathogenic hypotheses have been proposed for vitiligo, in part reflecting the incomplete understanding of the mechanisms that underlie this complex condition ( Fig. 66.1 ). A primary challenge is the fact that what is referred to as vitiligo likely represents a heterogeneous group of diseases with different genetic backgrounds and environmental triggers.
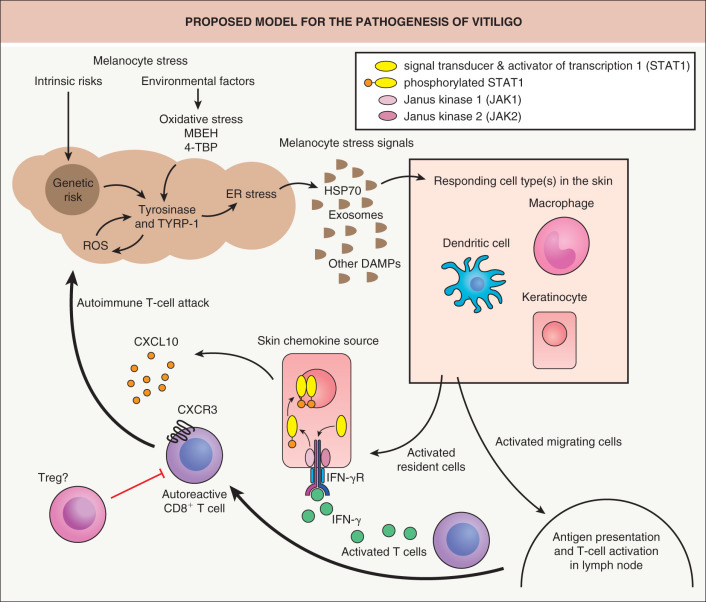
The immune system clearly plays a central role, in particular Th1 and Th17 cells, along with cytotoxic T cells, regulatory T cells, and dendritic cells; key mediators include interferon-γ (IFN-γ), C-X-C chemokine ligand 10 (CXCL10), and interleukin-22 (IL-22) . Animal models of vitiligo in which there are T cells reactive against melanocyte antigens have demonstrated the importance of an inflammatory dendritic cell (DC) phenotype and the INF-γ pathway .
Intrinsic defects of melanocytes and exogenous triggers may also play a role in vitiligo development. In addition, oxidative stress has been investigated as a pathogenic factor that could activate the immune response in vitiligo and underlie impaired WNT signaling that prevents melanoblast differentiation . Accumulating data highlight the complexity of vitiligo, with involvement of multiple cell types, including keratinocytes, fibroblasts, and stem cells as well as immune cells . These hypotheses are not mutually exclusive and the various pathways may converge to induce the disappearance of melanocytes from the skin and hair follicles. However, the exact cascade of events remains to be elucidated.
Clinical Features
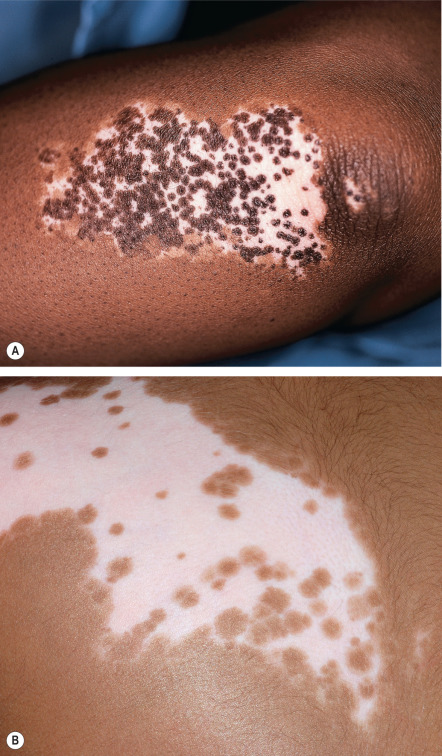
The most common presentation of vitiligo is totally amelanotic (milk- or chalk-white) macules or patches surrounded by normal skin. Well-developed lesions typically have discrete margins and may be round, oval, irregular, or linear in shape. The borders are usually convex, as if the depigmenting process were “invading” the surrounding normally pigmented skin. However, at their onset or when actively spreading, areas of vitiligo may be more ill-defined and hypo- rather than depigmented .
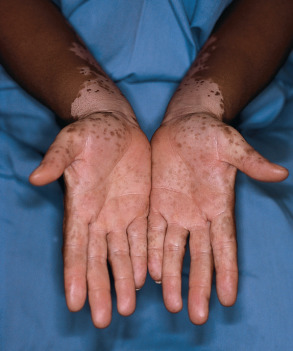
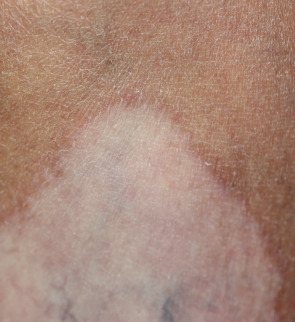
Lesions enlarge centrifugally over time at a rate than can be slow or rapid. Vitiligo macules and patches range from millimeters to centimeters in diameter and often have variable sizes within an area of involvement. In lightly pigmented individuals, the lesions may be subtle or inapparent without Wood’s lamp examination or tanning of uninvolved skin. In darkly pigmented patients, the contrast between vitiliginous areas and the surrounding skin is striking ( Fig. 66.2 ). Vitiligo is usually asymptomatic, but pruritus is occasionally noted, especially within active lesions.
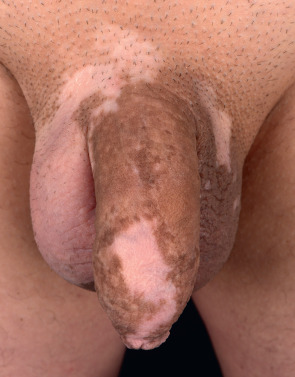
Vitiligo may develop anywhere on the body. Interestingly, it frequently localizes to sites that are normally relatively hyperpigmented, such as the face, dorsal aspect of the hands, nipples, axillae, umbilicus, and sacral, inguinal and anogenital regions (see Fig. 66.2 ). Typically, facial vitiligo occurs around the eyes and mouth (i.e. periorificial), and on the extremities it favors the elbows, knees, digits, flexor wrists, dorsal ankles and shins ( Figs 66.3 & 66.4 ). The most common sites of involvement are areas subjected to repeated trauma, pressure, or friction (e.g. in body folds or via contact with clothing). Palmoplantar and oral mucosal involvement in lightly pigmented individuals is often difficult to visualize without Wood’s lamp examination. In acrofacial vitiligo, periungual involvement of one or more digits may be associated with lip depigmentation; however, either can be an isolated finding.
The incidence of body leukotrichia varies from 10% to >60%, as vitiligo often spares follicular melanocytes. The occurrence of leukotrichia does not correlate with disease activity. Rarely, follicular vitiligo presents with leukotrichia in the absence of depigmentation of the surrounding epidermis . Although vitiligo of the scalp, eyebrows, and eyelashes usually presents as one or more localized patches of white or gray hair (poliosis; Fig. 66.5 ), scattered white hairs due to involvement of individual follicles or even total depigmentation of all scalp hair may occur.
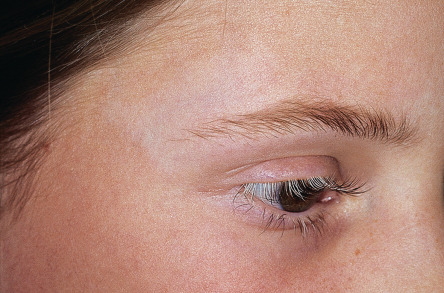
Clinical variants
Vitiligo ponctué , an unusual clinical presentation of vitiligo, is characterized by multiple, small, discrete amelanotic macules (confetti-like), sometimes superimposed upon a hyperpigmented macule. Erythema at the margin of a vitiligo macule is referred to as “vitiligo with raised inflammatory borders” or inflammatory vitiligo ( Fig. 66.6 ); a figurate papulosquamous variant has also been described. Occasionally, a hyperpigmented margin is seen. Blue vitiligo can result when vitiligo develops in areas of postinflammatory dermal pigmentation.
Trichrome vitiligo is characterized by a hypopigmented zone between the normal and depigmented skin. This intermediate zone has a fairly uniform hue rather than gradually progressing from white to normal. The number of melanocytes is also intermediate in this zone, suggesting a slower centrifugal progression compared with typical vitiligo. Quadrichrome and pentachrome vitiligo have also been described.
Hypochromic vitiligo (vitiligo minor) was recently described in patients with skin types V and VI . They presented with persistent hypopigmented macules in a seborrheic distribution, with lesions coalescing on the face and scattered on the neck, trunk, and scalp. A few individuals had additional achromic macules, and there was no history of prior inflammatory lesions. Decreased melanocyte density was noted histologically.
One of the manifestations of vitiligo is the isomorphic Koebner phenomenon (IKP), which is characterized by the development of vitiligo in sites of trauma, such as a surgical excision, burn, or abrasion. The IKP is more common in patients with progressive vitiligo, and it can occur in various forms of vitiligo. There appears to be a minimal threshold of injury required for the IKP to occur, bringing into question the hypothesis that minor trauma, such as friction from clothes (in the absence of true injury), can induce vitiligo lesions.
Clinical classification of vitiligo
Multiple attempts to classify the different types of vitiligo have resulted in confusing terminology. Two major forms are generally recognized: (1) segmental , which usually does not cross the midline; and (2) non-segmental , also simply called “vitiligo” without qualification . Mixed vitiligo refers to segmental and non-segmental forms occurring in the same patient .
The following classification scheme divides vitiligo into two major types, localized and generalized (see Fig. 66.4 ).
Localized:
- •
Focal : one or more macules in one area, but not clearly in a segmental distribution
- •
Unilateral/segmental : one or more macules involving one or rarely multiple segments of the body ( Fig. 66.7 ); lesions are typically unilateral and stop abruptly at the midline
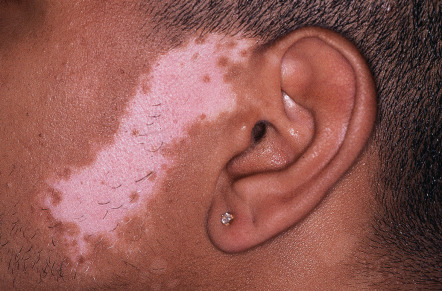
Fig. 66.7
Segmental vitiligo.
Unilateral band of depigmentation on the face, the most common location for segmental vitiligo. Note the pigmented and depigmented hairs within the affected area.
Courtesy, Kalman Watsky, MD.
- •
Mucosal : mucous membranes alone
- •
Vulgaris : scattered patches that are widely distributed
- •
Acrofacial : distal extremities and face
- •
Mixed : combination of segmental and generalized (acrofacial and/or vulgaris) types
- •
Universal : complete or nearly complete depigmentation
Course of the disease
The onset of vitiligo is usually insidious. Many patients become aware of the depigmented macules and patches, especially in sun-exposed areas, during the summer when tanning increases the contrast between involved and uninvolved skin. Clinical erythema or pruritus rarely precedes vitiligo.
The course of vitiligo is unpredictable. It becomes more extensive by the appearance of new depigmented macules, centrifugal enlargement of pre-existing lesions, or both processes. Peripheral hypopigmentation and poorly defined borders appear to be predictive of active vitiligo . A spotty pattern of depigmentation may also represent a marker of progressive disease. The natural course of generalized vitiligo is usually one of slow spread, but it may stabilize for a long period of time or evolve rapidly. Rarely, total body involvement develops within a few weeks or even days. In contrast, segmental vitiligo usually reaches its full extent within 1–2 years and remains restricted to the initial segmental area. The presence of halo nevi and leukotrichia increases the likelihood of evolution from segmental to mixed vitiligo . Some degree of sun-induced or spontaneous repigmentation of vitiligo is not uncommon, but complete and stable repigmentation is rare.
The v itiligo a rea s coring i ndex (VASI) and V itiligo E uropean T ask F orce (VETF) score represent validated quantitative assessment scales . However, universally accepted definitions for active and stable vitiligo are still lacking.
Vitiligo and ocular disease
The uveal tract (iris, ciliary body, and choroid) and retinal pigment epithelium contain pigment cells. Uveitis is the most significant ocular abnormality associated with vitiligo. Vogt–Koyanagi–Harada (VKH) syndrome is characterized by: (1) uveitis; (2) aseptic meningitis; (3) otic involvement (e.g. dysacusia); and (4) vitiligo, especially of the face or sacral region, and associated poliosis. Histologic examination of amelanotic skin, which classically appears after the extracutaneous symptoms, demonstrates an infiltrate consisting primarily of CD4 + lymphocytes, suggesting a prominent role for cell-mediated immunity.
Non-inflammatory depigmented lesions of the ocular fundus are evident in some patients with vitiligo, presumably representing focal areas of melanocyte loss. Although abnormal sensory hearing loss has been described in vitiligo patients, suggesting impairment of cochlear melanocytes, clearcut evidence of otic abnormalities remains to be demonstrated.
Alezzandrini syndrome is a rare disorder characterized by unilateral whitening of scalp hair, eyebrows, and eyelashes as well as ipsilateral depigmentation of facial skin and visual changes. In the affected eye, there is decreased visual acuity and an atrophic iris. The pathogenesis of Alezzandrini syndrome is unknown, but it is believed to be closely related to VKH syndrome.
Associated disorders
Although most vitiligo patients are otherwise healthy, generalized vitiligo is associated with a number of other autoimmune diseases, especially in patients with a family history of vitiligo and other forms of autoimmunity. Autoimmune thyroid disease occurs in ~15% of adults and ~5–10% of children with vitiligo , and other less frequently associated conditions include pernicious anemia, Addison disease, lupus erythematosus, rheumatoid arthritis, and adult-onset insulin-dependent diabetes mellitus . Of note, these conditions have been linked to the same autoimmunity predisposition genes as in vitiligo (e.g. PTPN22 , NLRP1 ; see Table 66.1 ). Halo melanocytic nevi, alopecia areata, and lichen sclerosus are additional autoimmune skin conditions that may be associated with vitiligo.
Patients with the autosomal recessive a utoimmune p oly e ndocrinopathy– c andidiasis– e ctodermal d ystrophy (APECED) syndrome often develop vitiligo. The gene responsible for APECED, AIRE ( a uto i mmune re gulator), encodes a transcription factor that promotes expression of tissue-specific self-antigens in the thymus, which facilitates development of peripheral tolerance. In APECED, failure to delete autoreactive T cells leads to autoimmune disease. In a mouse model, AIRE deficiency was found to result in tyrosinase-related protein-1 (TYRP1)-specific T cells that enhanced immune responses against melanoma .
Childhood vitiligo
Although vitiligo vulgaris is the most common clinical type observed in children, the frequency of segmental vitiligo (~15–30%) is significantly increased compared to that in adults (<5–10%) . The incidence of associated endocrinopathies is less than in the adult vitiligo population. A family history of vitiligo is associated with an earlier age of onset .
Pathology
In lesions of vitiligo, melanocytes are typically absent or present in very small numbers ( Fig. 66.8 ). The epidermal melanocyte density can be assessed with melanocyte-specific immunohistochemical stains, such as Melan-A (MART-1), MITF, and HMB45, or via incubation of biopsy specimens with dihydroxyphenylalanine (DOPA; detects tyrosinase activity). Although ultrastructural studies may be performed for research purposes, very few hypomelanotic disorders have specific ultrastructural findings.
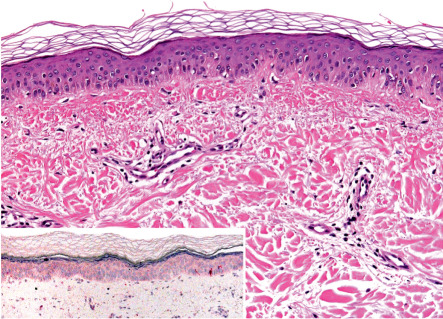
Differential Diagnosis
When there is complete depigmentation, the differential diagnosis may include chemical or drug-induced (e.g. imatinib) leukoderma, postinflammatory depigmentation, the leukodermas associated with melanoma and scleroderma, the late stages of treponematosis and onchocerciasis, and (for congenital lesions) piebaldism. A single circular depigmented lesion on the trunk of a young person may represent a stage III halo nevus (see below). Early lesions or those with a partial loss of pigment need to be distinguished from postinflammatory hypopigmentation, tinea versicolor, and other cutaneous infections (e.g. leprosy). In addition to a decrease, rather than an absence, of pigment, nevus depigmentosus can be distinguished by its stability and early onset, although lesions may not become apparent until mid-childhood in lightly pigmented individuals. Treatment with potent topical corticosteroids can also lead to hypomelanosis.
Treatment
The aims are stabilization of the depigmentation process and repigmentation. Although there is still no therapeutic panacea for vitiligo, available options can lead to satisfactory results. The therapeutic regimen depends on the extent, location, and activity of disease as well as the patient’s age, skin type, and motivation for treatment. In general, a period of at least 2–3 months is needed to determine whether a particular treatment is effective. The face, neck, mid extremities, and trunk tend to have the best response to therapy, while the distal extremities and lips are the most resistant to treatment. Repigmentation initially appears in a perifollicular pattern (unless the affected site is hairless or has depigmented hairs) and/or at the periphery of the lesions ( Fig. 66.9 ). After therapeutic repigmentation, the rate of recurrent depigmentation of vitiligo lesions is ~40% .
Corticosteroids
Topical corticosteroids are useful for localized areas of vitiligo. A meta-analysis showed that approximately half of patients with vitiligo affecting ≤20% of the body surface area (BSA) achieved >75% repigmentation with class 1 (superpotent) or 2–3 (high potency) topical corticosteroids; cutaneous atrophy was observed in 14% and 2% of these groups, respectively . To minimize side effects, class 1 corticosteroids can be used in 6–8-week cycles or on a twice-weekly basis, alternating with topical tacrolimus or a less potent topical corticosteroid . Treatment should be discontinued if there is no visible improvement after 2–3 months.
In general, intralesional corticosteroids should be avoided because of the pain associated with injection and the higher risk of cutaneous atrophy (≥30%). Systemic corticosteroid regimens utilizing high-dose pulses, mini-pulses, or low daily oral doses have been reported to arrest rapidly spreading vitiligo and induce repigmentation . However, given the potential for serious side effects, the role of systemic corticosteroids in the treatment of vitiligo remains controversial.
Topical calcineurin inhibitors (TCIs)
Multiple studies have shown that topical tacrolimus 0.1% ointment or pimecrolimus 1% cream applied twice daily can result in repigmentation of vitiligo, with response rates in pediatric patients similar to those achieved with topical corticosteroids. The best results are obtained when these agents are used on the face and other sun-exposed areas, suggesting a synergistic effect. TCIs can also enhance repigmentation when used in conjunction with narrowband UVB phototherapy or the excimer laser . Of note, avoidance of UV light is suggested by the package insert for TCIs, and the risk:benefit ratio should be discussed. Topical tacrolimus can also be used together with topical corticosteroids on a rotational basis (see above) or as maintenance therapy after repigmentation. In a recent randomized controlled study, bi-weekly application of tacrolimus 0.1% ointment to sites of previous vitiligo reduced the rate of recurrent depigmentation to 10%, compared to 40% with placebo .
Photo(chemo)therapy
Narrowband UVB
Narrowband UVB (NB-UVB; see Ch. 134 ) has become the first-line treatment for adults and children ≥6 years of age with generalized vitiligo , especially if it involves ≥20% of the body surface area or cosmetically sensitive areas that typically respond to treatment. Several studies have demonstrated the effectiveness of NB-UVB monotherapy , which is superior to broadband UVB for the treatment of vitiligo. The starting dose of NB-UVB ranges from 100–250 mJ/cm 2 , which is increased in increments of 10–20% at each subsequent exposure until mild, asymptomatic lesional erythema is achieved. Treatments are administered 2–3 times per week, but not on two consecutive days . Short-term side effects include pruritus and xerosis; long-term side effects (e.g. cutaneous carcinogenesis) have not been reported. The advantages of NB-UVB over psoralen plus UVA (PUVA) therapy include shorter treatment times, no gastrointestinal side effects, reduced phototoxic reactions, less accentuation of the contrast between depigmented and normally pigmented skin, and no need for post-treatment photoprotection. In addition, NB-UVB can be used in children, pregnant or lactating women, and individuals with hepatic or kidney dysfunction.
Psoralen plus UVA
Psoralen photochemotherapy involves the use of psoralens combined with UVA light (see Ch. 134 ). The psoralen most commonly used is 8-methoxypsoralen (8-MOP, methoxsalen). 5-methoxypsoralen (5-MOP, bergapten) is not approved in the US and 4,5′,8-trimethylpsoralen (TMP, trioxsalen) is no longer commercially available. Psoralens can be administered orally (oral PUVA) or applied topically (topical PUVA), followed by exposure to either UVA light or natural sunlight (PUVASOL).
Oral PUVA treatments using 8-MOP (0.4–0.6 mg/kg) are typically administered two times weekly. For patients with vitiligo, the initial dose of UVA is usually 0.5–1.0 J/cm 2 , which is gradually increased until minimal asymptomatic erythema of the involved skin occurs. To reduce the risk of the Koebner phenomenon, significant erythema (phototoxicity) is avoided. 5-MOP has about the same response rate as 8-MOP in repigmenting vitiligo, but a lower incidence of phototoxicity as well as less nausea and vomiting.
The response rate to PUVA is variable; it often produces cosmetically acceptable improvement, but complete repigmentation is uncommon. The total number of PUVA treatments required is generally 50–300. The absolute and relative contraindications, as well as the short- and long-term side effects, of oral PUVA therapy are reviewed in Chapter 134 . To date, only a few vitiligo patients with PUVA-induced cutaneous carcinomas have been reported. Although this probably reflects a smaller cumulative UVA dose than in patients treated for other disorders such as psoriasis, large follow-up studies have not yet been done in PUVA-treated vitiligo patients. Until more data are available, it seems wise to recommend 1000 J/cm 2 as the maximum cumulative (P)UVA dose and 300 as the maximum number of UVA treatments.
Topical (paint) PUVA is more difficult to perform because of the high risk of phototoxicity and subsequent blistering or koebnerization. A low concentration (≤0.1%) of psoralen should be used, which requires dilution of the commercially available preparation. Approximately 20–30 minutes after applying the topical cream or ointment onto the lesions, the patient should be exposed to initial UVA doses of no more than 0.25 J/cm 2 , with the same fractional increments until mild erythema is achieved in the treated sites.
PUVASOL (psoralens + natural sunlight) can be used in sunnier climates, utilizing the same principles as for PUVA. Less phototoxic oral psoralens such as 5-MOP are preferred in order to avoid phototoxic reactions.
Other phototherapies
Oral khellin plus UVA (KUVA) and phenylalanine plus UVA have also been employed. There have been conflicting reports regarding efficacy as well as concerns regarding the hepatotoxicity of khellin. As a result, these modalities are not recommended. Topical KUVA has a relatively low risk of phototoxicity, but it requires a longer duration of treatment and higher UVA doses than does oral PUVA .
Focused microphototherapy has the advantage of irradiating only the depigmented skin. A directed beam of broadband or narrowband UVB light is applied to areas of vitiligo using spot sizes of 1–5 cm. Treatments are administered from several times weekly to twice monthly. In one large study, 70% of patients who received a mean of 24 treatments over a 12-month period achieved >75% repigmentation.
Lasers and related light devices
Excimer laser and lamp
The operational wavelength of the 308 nm excimer laser and lamp is close to that of NB-UVB . The therapeutic benefit of the excimer laser for vitiligo has been investigated in multiple studies, and, overall, 20–50% of lesions achieve ≥75% repigmentation ; the excimer lamp appears to have similar efficacy . Only a few studies have directly compared excimer laser to NB-UVB, and some but not all showed superior results with the former modality . This may be explained by the excimer laser’s higher irradiance (power per unit area), which is thought to stimulate melanocyte development .
Localized patches of vitiligo are treated one to three times weekly with the excimer laser, typically for a total of 24 to 48 sessions; the repigmentation rate depends on the total number of sessions, not their frequency . In practice, twice weekly treatments for a total of ~40 sessions is thought to be optimal. As with other vitiligo therapies, facial lesions respond better than those on the distal extremities and overlying bony prominences . Erythema and (rarely) blistering represent potential side effects .
Helium–neon laser
The helium–neon laser emits a wavelength (632.8 nm) in the red visible light range that can enhance melanocyte proliferation and melanogenesis in vitro . In a study performed in 30 patients with segmental vitiligo, 20% of lesions achieved ≥75% repigmentation after a mean of 79 treatment sessions, which were administered once or twice weekly .
Surgical therapies
For vitiligo patients who fail to respond to medical therapy, surgical treatment with autologous transplantation techniques may be an option . The general selection criteria for autologous transplantation include stable disease for ≥6 months, absence of the Koebner phenomenon, no tendency for scar or keloid formation, and age >12 years . A minigraft test showing retention/spread of pigment at the recipient site and no koebnerization at the donor site after 2–3 months can also assist in patient selection.
Several methods of surgical repigmentation have been successfully utilized. Minigrafting is the simplest technique. Small punch grafts (1–2 mm) from uninvolved skin are implanted within achromic areas, separated from each other by 5–8 mm. A cobblestone effect, a variegated appearance of the grafts, and sinking pits represent potential unfavorable outcomes. Because scarring and dyspigmentation may occur at the donor sites, cosmetically insensitive areas are chosen. The advantages of suction blister epidermal grafting are the absence of scarring at the donor site and the possibility of reusing this area. However, failure of the graft to take and koebnerization may occur. Grafting of cultured autologous melanocytes is an expensive technique that requires specialized laboratory expertise; grafts consist of pure melanocytes or melanocytes admixed with keratinocytes . To avoid the need for in vitro culture, which involves mitogens to enhance cell growth, grafting of non-cultured epidermal cell suspensions that include melanocytes has been advocated . Grafting of individual hairs to repigment vitiligo leukotrichia has also been successfully performed. Most of these techniques require clinical expertise.
Combination therapy
Combination therapy may produce higher rates of repigmentation compared to traditional monotherapies. Examples include phototherapy following surgical procedures as well as combining TCIs and/or topical corticosteroids with NB-UVB or excimer laser therapy . Although topical vitamin D derivatives are relatively ineffective as monotherapy, these agents may result in additional repigmentation when used in conjunction with phototherapy.
Micropigmentation
The technique of permanent dermal micropigmentation utilizes a non-allergenic iron oxide pigment to camouflage recalcitrant areas of vitiligo. This tattooing method is especially useful for the lips, nipples and distal fingers, which have a poor rate of repigmentation with currently available treatments. Although the color may not match perfectly with the normal surrounding skin and can fade over time, the result is immediate and can represent a dramatic aesthetic improvement.
Depigmentation
Depigmentation represents a treatment option for patients who have widespread vitiligo with only a few areas of normally pigmented skin in exposed sites. The patients must be carefully chosen, i.e. adults who recognize that their appearance will be altered significantly and who understand that depigmentation requires lifelong strict photoprotection (e.g. sunscreens, clothing, umbrellas). The most commonly used agent is 20% monobenzyl ether of hydroquinone (MBEH), applied once to twice daily to the affected areas for 9–12 months or longer . MBEH is a potent irritant and allergen, and an open application test can be performed before more widespread application. It typically takes 1–3 months to initiate a response, and a loss of pigment can occur at distant sites. Although depigmentation from MBEH is considered permanent, repigmentation (especially perifollicular in areas with pigmented hairs) can be seen following a sunburn or intense sun exposure. Monomethyl ether of hydroquinone (MMEH) in a 20% cream can be used as an alternative to MBEH . Side effects include contact dermatitis, exogenous ochronosis, and leukomelanoderma en confetti. Depigmentation via Q-switched ruby laser therapy was reported to achieve faster depigmentation than that achieved with a bleaching agent , and this laser has also been used in combination with topical 4-methoxyphenol to induce depigmentation . Lastly, depigmentation with the Q-switched alexandrite laser has been described .
Psychological support
The impact of vitiligo on quality of life is severe in many affected individuals, and it is critical for physicians to recognize this aspect of the condition and address their patients’ psychological needs. Although a “magic” treatment is not yet available, there is always something beneficial that can be done for vitiligo patients. They first need to know what their skin disorder is. Explaining the nature of the disease process and the potential and limits of available therapies is important and more productive than a fatalistic attitude that there is no cure and vitiligo is “only” a cosmetic disorder. Even helping patients to conceal the condition so that it is not visible can be part of the management plan. The use of support groups and, if indicated, psychological counseling are important supplementary therapies.
Additional controversial therapies
Pseudocatalase with narrowband UVB
The rationale for this treatment is based on the hypothesis that accumulation of hydrogen peroxide leads to pathogenic inactivation of catalase in the skin of patients with vitiligo ( Table 66.2 ). In an open uncontrolled study, complete repigmentation of lesions on the face and hands was observed in 90% of patients (30 of 33) treated with topical pseudocatalase and calcium twice daily plus UVB twice weekly, with initial repigmentation at 2–4 months . Controlled trials, however, showed no efficacy of topical pseudocatalase/superoxide dismutase compared to placebo and no additional benefit of pseudocatalase compared to UVB alone .
| PATHOGENIC HYPOTHESES FOR VITILIGO | ||
| Hypothesis | Evidence for hypothesis | Evidence against hypothesis |
| Autoimmune destruction of melanocytes |
|
|
| An intrinsic defect in melanocytes, their adhesive properties, and/or factors critical to their survival |
|
|
| Defective defense against oxidative stress leading to destruction of melanocytes |
|
|
Systemic antioxidant therapy
The rationale for this approach rests on the hypothesis that vitiligo results from a deficiency of natural antioxidant mechanisms. Although to date not validated by controlled clinical trials, selenium, methionine, tocopherols, ascorbic acid, and ubiquinone are prescribed by some physicians.
Potential emerging treatments
Ablative laser treatment followed by narrowband UVB plus topical 5-fluorouracil or corticosteroids
In difficult-to-treat sites (e.g. distal extremities, over bony prominences), erbium:YAG laser ablation of vitiligo lesions followed by NB-UVB therapy twice weekly for 3–4 months, plus topical application of either 5-fluorouracil or a potent corticosteroid, was found to result in significantly greater repigmentation than treatment with NB-UVB ± the topical corticosteroid . Similar studies have shown that treatment of recalcitrant vitiligo lesions with an ablative fractional carbon dioxide laser led to greater efficacy of subsequent therapy with NB-UVB, outdoor sun, and/or a potent topical corticosteroid . In order to minimize koebnerization, these approaches should be reserved for patients with stable vitiligo.
Topical prostaglandins
Preliminary studies have suggested the utility of topical prostaglandin E 2 and latanoprost (an analogue of prostaglandin F 2 ) in the treatment of vitiligo . Although interesting, these results require confirmation.
Afamelanotide
Afamelanotide is an α-melanocyte stimulating hormone (α-MSH) analogue that stimulates melanogenesis and melanocyte proliferation by binding to the melanocortin-1 receptor (MC1R; see Ch. 65 ). A recent randomized controlled study found that the addition of afamelanotide (monthly subcutaneous implants) to NB-UVB therapy increased the speed and extent of repigmentation compared to NB-UVB alone in patients with generalized vitiligo, especially those with skin types IV–VI . However, afamelanotide-induced excessive tanning of non-lesional skin can increase the contrast with lesional skin, thereby reducing cosmetic acceptance in lightly pigmented patients . Additional studies are needed to determine the indications and limitations of afamelanotide therapy for vitiligo.
Janus kinase (JAK) inhibitors
Administration of the JAK inhibitors ruxolitinib and tofacitinib has been reported to lead to repigmentation of vitiligo. Further investigation is needed to determine how approaches targeting the IFN-γ–JAK–STAT1 signaling pathway that drives melanocyte destruction can be utilized to treat vitiligo (see Fig. 66.1 ).
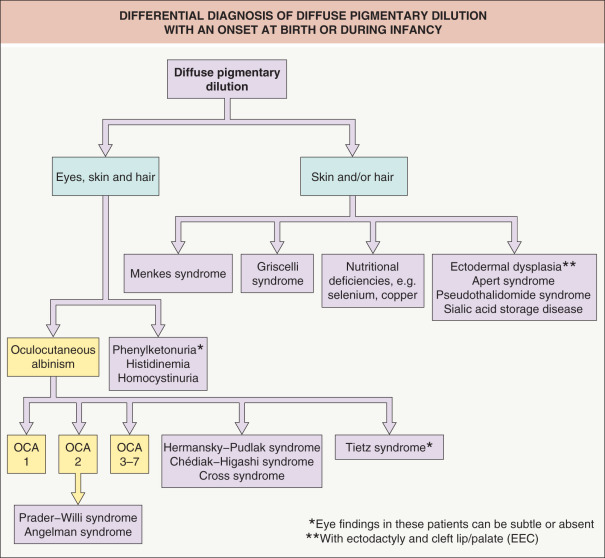
Hereditary Hypomelanosis
Oculocutaneous Albinism
Oculocutaneous albinism (OCA) consists of a group of genetic disorders characterized by diffuse pigmentary dilution due to a partial or total absence of melanin pigment within melanocytes of the skin, hair follicles, and eyes . The number of epidermal and follicular melanocytes is normal. Hypopigmentation involving primarily the retinal pigment epithelium is termed ocular albinism (OA).
Epidemiology
OCA is the most common inherited disorder that leads to diffuse hypomelanosis. In most populations, the estimated frequency is 1 : 20 000; however, it is as high as 1 : 1500 in some African tribes.
Pathogenesis
All well-characterized types of OCA have an autosomal recessive inheritance pattern, although a few rare families with autosomal dominant OCA have been described . Based upon molecular studies, four types of OCA have been defined (see Table 65.1 ). OCA type 1 (OCA1) results from reduced (OCA1B) or absent (OCA1A) tyrosinase activity; at least 320 distinct mutations in the tyrosinase gene ( TYR ) have been identified in patients with OCA1 . OCA2 is due to mutations in the P gene (pink-eyed dilution; now referred to as OCA2 ) . Although the function of the P protein is still debated, studies have pointed to a possible role in regulating organelle pH and facilitating vacuolar accumulation of glutathione.
OCA3 results from mutations in the tyrosinase-related protein 1 ( TYRP1 ) gene . The TYRP1 protein is a melanocyte-specific gene product involved in eumelanin synthesis (see Fig. 65.11 ), probably via stabilization of tyrosinase. Both OCA1 and OCA3 appear to be endoplasmic reticulum (ER) retention diseases wherein the abnormal proteins (tyrosinase or TYRP1) never leave the ER to become incorporated into melanosomes (see Fig. 65.7 ). Of note, dysfunction of the P protein can also lead to abnormal processing and trafficking of tyrosinase. OCA4 is caused by mutations in SLC45A2 which encodes solute carrier family 45 member 2 (formerly membrane-associated transporter protein [MATP]), a transmembrane transporter with roles in tyrosinase processing and intracellular trafficking of proteins to the melanosome .
OCA5 has been linked to chromosome 4q24, but the responsible gene has not yet been identified. OCA6 was recently found to result from mutations in SLC24A5 , which encodes a putative cation exchanger localized to the melanosomal membrane . Of note, this gene was previously identified as one of the determinants of the physiologic variation in human pigmentation. Lastly, mutations in C10orf11 (chromosome 10 open reading frame 11) underlie OCA7; the C10orf11 protein is expressed in melanoblasts as well as melanocytes and is thought to have a role in melanocyte differentiation .
Autosomal recessive OA (AROA) is genetically heterogeneous, and some cases actually represent OCA1B or OCA2 but with subtle cutaneous findings. The most common form of OA is an X-linked recessive disorder caused by mutations in the G protein-coupled receptor 143 gene ( GPR143 ; formerly OA1 ), which encodes a pigment cell-specific intracellular G protein-coupled receptor that regulates melanosome formation and transport within melanocytes and cells of the retinal pigment epithelium . Mutations in GPR143 that lead to ocular albinism can result in retention of the aberrant protein within the ER.
Clinical features
Ocular manifestations
The many ocular manifestations of OA and OCA reflect a reduction in melanin within eye structures or misrouting of optic nerve fibers during development. The former leads to a translucent iris that transmits light upon globe transillumination, as well as a relatively hypopigmented retina and fovea that are associated with photophobia and reduced visual acuity; the severity of these findings correlates with the amount of reduction in melanin pigment. Misrouting of the optic fibers is thought to be responsible for the characteristic strabismus, nystagmus, and lack of binocular vision.
OCA1
There are two clinical subtypes of OCA1, based upon whether tyrosinase activity is reduced (OCA1B) or absent (OCA1A).
OCA1A
OCA1A corresponds to the classic “tyrosinase-negative” OCA. The melanocytes of the skin, hair, and eyes synthesize no melanin. The characteristic phenotype includes white hair, milky white skin, and blue–gray eyes at birth. With age, the skin color remains white and melanocytic nevi amelanotic, but the hair may develop a slight yellow tint due to denaturing of hair keratins. These patients have an extreme sensitivity to UV light and a strong predisposition to skin cancer. Reduced visual acuity is most severe in OCA1A, and some patients are legally blind.
OCA1B
Because the decrease in tyrosinase activity varies, the phenotype ranges from obvious to subtle pigmentary dilution when compared with first-degree relatives. One of the original OCA1B phenotypes was called “yellow albinism” because of the eventual color of the patient’s hair, since the formation of yellow pheomelanin requires less tyrosinase activity. Other clinical types of OCA1B have been referred to as “minimal pigment OCA”, “platinum OCA”, and “temperature-sensitive OCA”. All of these patients have little or no pigment at birth, but they develop some pigmentation of the hair and skin during the first and second decades of life. The majority burn without tanning after sun exposure, and some degree of iris translucency is often present. Amelanotic or pigmented melanocytic nevi can develop.
In the temperature-sensitive OCA1B phenotype, patients are born with white hair and skin and blue eyes. During puberty, scalp and axillary hairs remain white, but arm hairs turn light reddish brown and leg hairs turn dark brown. The abnormal tyrosinase enzyme is temperature-sensitive, losing its activity above 35°C. As a result, melanin synthesis does not occur in warmer areas of the body, akin to the phenotype of a Siamese cat, which also has temperature-sensitive tyrosinase activity.
OCA2
The OCA2 phenotype (in addition to OCA1B) corresponds to the classic “tyrosinase-positive” OCA. The clinical spectrum of OCA2 is broad, ranging from minimal to moderate pigmentary dilution of the hair, skin, and iris, with little to no ability to tan. The vast majority of individuals of African descent who have “tyrosinase-positive” OCA have OCA2. With time, pigmented melanocytic nevi and lentigines may develop in sun-exposed areas ( Fig. 66.10 ); the latter can become rather large and darkly colored ( Fig. 66.11 ). Another phenotype called “brown OCA” in African and African-American populations results primarily from mutations in the P gene. In these individuals, the hair and skin are light brown, the irides are gray to tan at birth, and sunburns are unusual.
The hypopigmentation seen in a subset of patients with Prader–Willi syndrome (PWS) and Angelman syndrome (AS) represents a form of OCA2. PWS features hyperphagia, obesity, hypogonadism, and intellectual disability, whereas AS is characterized by severe intellectual disability, microcephaly, ataxic movements, and inappropriate laughter. Due to genomic imprinting (see Ch. 54 ), deletions of the 15q region (which includes the P gene) in the paternal chromosome lead to PWS, while deletions in the maternal chromosome give rise to AS. Approximately 1% of patients with AS or PWS also have OCA2, which occurs when deletion of one copy of the P gene is accompanied by a mutation in the second copy.
OCA3
The phenotypes of individuals with OCA3 are classified as “rufous” (vast majority of OCA3 patients) and “brown” (more often seen in OCA2). Rufous OCA has been identified in individuals with type III–V skin color, and the phenotype includes a red–bronze skin color, ginger-red hair, and blue or brown irides. Rufous OCA is associated with mutations in TYRP1 . A patient with the brown OCA phenotype due to mutations in both copies of TYRP1 has also been described: an African-American child with light brown skin, light brown hair and blue–gray irides.
OCA4
OCA4 is most common among individuals with albinism who are from Japan (~25% of patients), China (10–20% of patients), or India (~10% of patients), and it accounts for ~5% of albinism in Caucasians . The phenotype of OCA4 is variable; hair color ranges from white to yellow or brown, and patients may or may not develop increased pigmentation of the skin and hair over time.
OCA5–7
OCA5 was described in a consanguineous Pakistani family, in which affected individuals had white skin and golden hair. Patients with OCA6 from China and French Guiana were said to have white to light brown skin with some ability to tan, yellow hair at birth that darkened with age, and light brown irides . OCA7 has been described primarily in patients from the Faroe Islands of Denmark; affected individuals have lighter skin than their relatives and hair color ranging from light yellow to brown .
Ocular albinism type 1 (OA1)
Ocular albinism type 1 is characterized by a substantial reduction in visual acuity, hypopigmentation of the retina, and the presence of macromelanosomes in the eyes. Affected boys have nystagmus, photophobia, and foveal hypoplasia . Their skin is usually clinically normal without notable pigmentary dilution, although hypopigmented macules have been described in affected individuals who have darkly pigmented skin. Macromelanosomes are evident on histologic examination of the skin.
Pathology
Although there is a reduction in melanin content, a normal number of melanocytes are present within the epidermis.
Differential diagnosis
This is outlined in Fig. 66.12 . Occasionally, patients with total body vitiligo may be thought to have OCA, but their epidermis lacks melanocytes.