The term vasculitis refers to inflammation of blood vessels, a finding that can be seen in association with a variety of clinical manifestations and as either a primary or secondary phenomenon. Along with vessel inflammation there is deposition of fibrinoid material in vessel walls and the presence of nuclear fragments (nuclear dust) resulting from disintegration of neutrophilic nuclei (karyorrhexis) within the vessel wall and surrounding tissues (hence the terminology leukocytoclastic vasculitis ). The presence of vasculitis may result in tissue injury owing to vascular stenosis, occlusion, aneurysm, or rupture. The incidence of childhood vasculitis is approximately 50 cases per 100,000 children per year, with geographic differences noted throughout the world.
The vasculitic disorders are a heterogeneous group of conditions, many of which present with (or eventually result in) cutaneous manifestations. Classification of the vasculitides is difficult and has most often been based on the gross and microscopic features of the disease process, primarily the vessel size involved (i.e., large, medium-sized, or small). Large-vessel vasculitides, which will not be discussed here, include giant-cell (temporal) arteritis and Takayasu arteritis. Medium-sized vasculitides involve predominantly visceral arteries and include two of the most common forms of pediatric vasculitis syndromes, polyarteritis nodosa (PAN) and Kawasaki disease (KD), both of which will be discussed. The other entities covered in this chapter all represent small-vessel disorders, which tend to involve primarily capillaries and venules. It should be remembered that this classification of vasculitis based on vessel size is imprecise, and overlap in the size of involved vessels is common. A summary classification of pediatric vasculitides is shown in Table 21-1 . The original patient groups used to develop vasculitis classification criteria by the American College of Rheumatology (ACR) did not include children. Subsequently the vasculitis working group of the Pediatric Rheumatology European Society (PRES), in conjunction with the European League Against Rheumatism (EULAR) and the Pediatric Rheumatology International Trials Organization (PRINTO), more recently developed and validated criteria for some of the most common childhood vasculitides, a process that culminated at the 2008 Ankara Consensus Conference. A summary of these criteria is shown in Table 21-2 .
| Predominant Vessel Size | Disorder |
|---|---|
| Large | Takayasu arteritis |
| Medium | Polyarteritis nodosa |
| Kawasaki disease | |
| Small | Hypersensitivity vasculitis (cutaneous leukocytoclastic vasculitis) |
| Henoch–Schönlein purpura | |
| Acute hemorrhagic edema of infancy | |
| Hypocomplementemic urticarial vasculitis | |
| Cryoglobulinemic vasculitis | |
| Erythema elevatum diutinum (primarily adults) | |
|
| Disorder | Diagnostic Criteria |
|---|---|
| Henoch–Schönlein purpura |
|
| Childhood polyarteritis nodosa | |
| Granulomatosis with polyangiitis ‖ |
* Also referred to as EULAR/PRINTO/PRES criteria .
† For purpura with atypical distribution, demonstration of IgA deposit in biopsy required.
‡ Angiography: aneurysm, stenosis, or occlusion of medium- or small-sized artery.
§ Livedo reticularis, nodules, infarctions.
‖ Formerly known as Wegener granulomatosis.
¶ Purulent/bloody nasal discharge, recurrent epistaxis/crusts/granulomas, septal perforation or saddle nose deformity, sinusitis.
Henoch–Schönlein (Anaphylactoid) Purpura
Henoch–Schönlein purpura (HSP), also known as anaphylactoid purpura , is a form of small-vessel vasculitis that occurs primarily in children (especially boys) between 2 and 11 years of age, most commonly occurring in children under the age of 5 years. It is the most common type of childhood systemic vasculitis. The classic presentation is a combination of nonthrombocytopenic palpable purpura in dependent areas, arthritis, abdominal pain, and glomerulonephritis. HSP is an inflammatory disorder that has been linked hypothetically to several potential etiologic agents including group A β-hemolytic streptococci (GABHS), other bacterial or viral organisms, immunizations, and drugs, although the exact etiology remains unclear. There are reports of patients with coexisting HSP and acute rheumatic fever, highlighting the potential association between GABHS and HSP. Bartonella henselae has also been suggested as a potential etiologic infectious agent. Although the nature of the immunologic reaction in HSP is not completely clear, the common history of antecedent upper respiratory tract infection preceding the onset of symptoms suggests a hypersensitivity phenomenon resulting in localized or widespread vascular damage. A fairly consistent immunologic observation is the deposition of immunoglobulin (Ig) A immune complexes in affected organs (i.e., skin, kidneys) when studied by immunofluorescence. IgA complexes, however, are not a necessary requirement for the development of HSP, and their absence should not exclude the diagnosis. Around one-third of patients have elevated serum IgA levels. Several polymorphisms involving cytokines and cell-adhesion molecules that modulate inflammatory responses and endothelial cell activation have been observed and may correlate with disease susceptibility, extent of involvement, and/or severity of renal disease. Mutations in the MEFV gene, which is classically associated with familial Mediterranean fever with homozygous or compound heterozygous mutation, have been described in some patients with HSP and may portend more common gastrointestinal and joint involvement and edema.
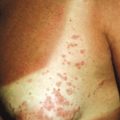
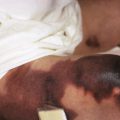
The clinical picture of HSP is often, but not always, distinctive. Most patients with a rash present with an initial urticarial eruption of macules and papules ( Fig. 21-1 ) that rapidly become purpuric and are distributed primarily on the lower extremities ( Figs. 21-2 through 21-4 ) and buttocks ( Fig. 21-5 ). Lesions may also occur on the upper extremities, trunk, and face, and in some instances skin lesions may develop in patterns at pressure sites. In some patients, petechiae ( Fig. 21-6 ) or ecchymoses (“bruises”) ( Fig. 21-7 ) may predominate over palpable purpuric lesions. The cutaneous eruption is the presenting feature of HSP in 50% of cases. Newer purpuric lesions develop and may result in a polymorphous appearance. Other skin lesion morphologies, including vesicular or bullous lesions, erosions or ulcers, necrosis, gangrene, and even erythema multiforme-like lesions, may occur. Marked edema of the hands, feet, scalp, or face may also be seen, especially in younger patients. The disease often consists of a single episode that may last for several days to weeks, but in some cases recurrent attacks may occur at intervals for weeks to months.
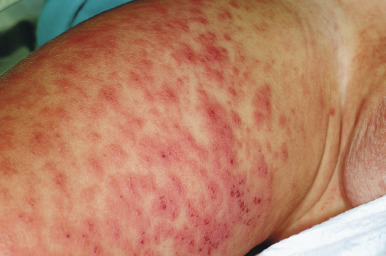
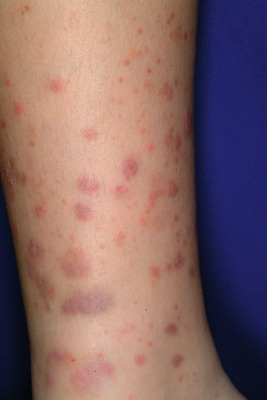
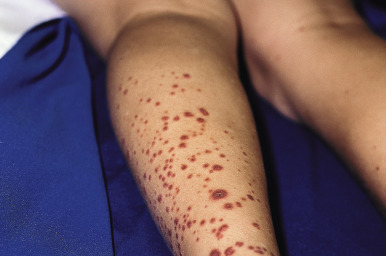
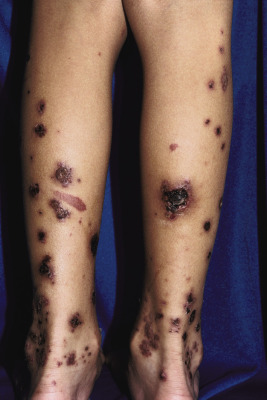
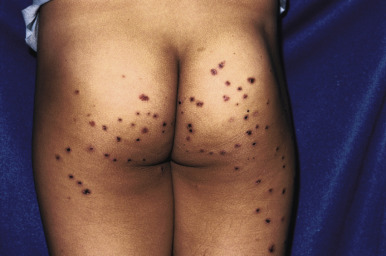
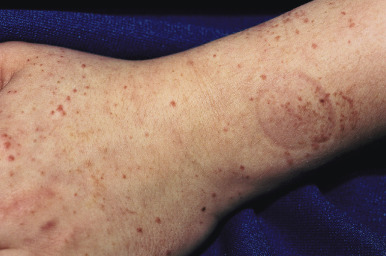
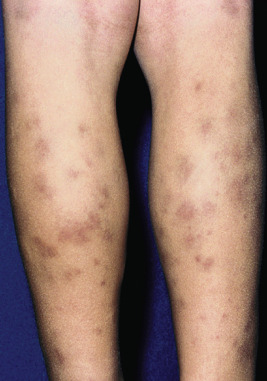
Individual lesions of HSP occur in crops, tend to fade after about 5 days, and eventually are replaced by areas of hyperpigmentation or ecchymoses. Although the differential diagnosis of the skin lesions may include drug reaction, erythema multiforme, or urticaria, the presence of palpable purpura (the hallmark of leukocytoclastic vasculitis) will usually clarify the true nature of the disorder. This characteristic finding, created by edema and extravasation of erythrocytes, gives individual lesions their diagnostic palpable and purpuric appearance as well as their nonblanching quality (i.e., individual lesions cannot be blanched when viewed through a glass slide exerting pressure over the surface, a technique called diascopy ).
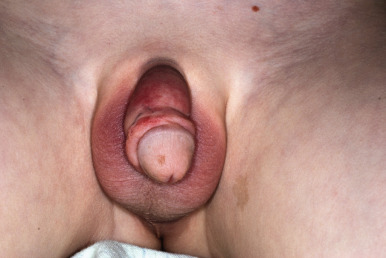
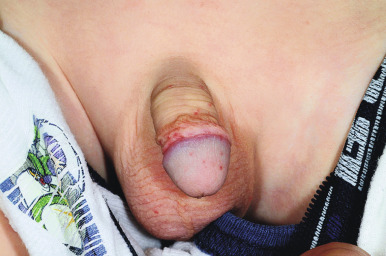
Occasionally the face, mucous membranes of the mouth and nose, and the anogenital regions may show petechiae. In males, vasculitis of the scrotal vessels may lead to acute scrotal swelling with or without erythema or purpura ( Fig. 21-8 ). Pain may be severe, and this presentation may mimic that of testicular torsion or other causes of “acute scrotum,” necessitating ultrasonography and/or radionuclide scans to help differentiate between these disorders. In one study, roughly 20% of males with HSP had scrotal involvement, and these patients were noted to have an association with headache, edema, and elevated C3 levels.
Systemic involvement in HSP, which is seen in up to 80% of cases, most commonly occurs in the kidneys, gastrointestinal tract, and joints. The degree of systemic involvement varies, with joint or gastrointestinal symptoms seen in as many as two-thirds of affected children. Renal disease occurs most often and is the most significant correlate of long-term prognosis.
The clinical expression of HSP kidney involvement varies from transient microscopic hematuria to rapidly progressive glomerulonephritis. Glomerulonephritis is seen overall in 40% to 50% of children with HSP and is usually transient and benign. The renal lesion in HSP is indistinguishable histopathologically from that of IgA nephropathy (Berger disease), although the latter tends to occur more often in older patients. The renal involvement may not become apparent for several weeks; hence the importance of long-term follow-up monitoring for development of this complication. Most often, however, if kidney involvement is going to be seen, it will be found within 3 months of disease onset. Potential correlates of kidney disease in HSP include age over 4 years, persistent purpura, severe abdominal pain, and depressed coagulation factor XIII activity less than 80%. Initial renal insufficiency appears to be the single best predictor of the further clinical course in children with HSP nephritis.
Gastrointestinal signs and symptoms result from edema and hemorrhage of the bowel wall as a result of vasculitis. They most often include colicky abdominal pain (that may be severe and associated with vomiting), hematochezia, and hematemesis. Visceral infarction or perforation, pancreatitis, cholecystitis, esophageal involvement, colitis, protein-losing enteropathy, intussusception, hemorrhage, or shock may also be associated. Intussusception, seen in up to 2% of patients, is more common in boys. Because non-HSP-associated intussusception generally occurs in children younger than 2 years of age, when intussusception is diagnosed in older children HSP must be strongly considered. Chronic intestinal obstruction with ileal stricture has also been reported, occurring months after resolution of the acute illness.
Joint involvement in HSP is characterized by tender and painful joints with primarily periarticular swelling. True arthritis is less common. Although the ankles and knees are most often affected, the elbows, hands, and feet may also be involved. HSP joint symptoms tend to be oligoarticular. Arthritic symptoms are usually transient and rarely result in permanent deformity.
In addition to the skin, genitourinary and gastrointestinal tracts, and joints, other organ involvement is occasionally seen in HSP. Central nervous system (CNS) involvement results most commonly in headache and occasionally behavioral alteration, hyperactivity, intracerebral bleeds, paresis, or seizures. Status epilepticus has been rarely reported. Respiratory involvement may range from an asymptomatic pulmonary infiltrate to recurrent episodes of pulmonary hemorrhage. Bleeding diathesis may occasionally occur.
The diagnosis of HSP is seldom difficult when patients exhibit the classic “tetrad” of organ involvement (skin, kidneys, gastrointestinal tract, and joints). However, many patients show only some of these findings, and the differential diagnosis depends on which organ-specific symptoms predominate. The differential diagnosis of palpable purpura may include hypersensitivity vasculitis (HV) (usually caused by drug reaction; see Hypersensitivity Vasculitis section), hemorrhagic diathesis (i.e., factor deficiency), or sepsis. In addition, the more “ecchymotic” lesions of HSP may be mistaken for child abuse and must be evaluated within the context of the overall clinical presentation. When the diagnosis remains in doubt, histopathologic examination of a skin biopsy specimen is useful in confirming the presence of vasculitis. Direct immunofluorescence revealing IgA immune complexes is highly suggestive of but not pathognomonic for HSP. Other laboratory investigations to be considered include serum chemistry profile, blood-cell counts, coagulation studies, abdominal radiographic studies, stool guaiac testing, urinalysis, and kidney biopsy.
The overall prognosis for most patients with HSP is excellent, and full recovery without permanent residua is the norm. The disease tends to run its course over 4 to 6 weeks. In younger children, the disease is generally milder and of shorter duration with fewer renal and gastrointestinal manifestations and few recurrences. Renal disease is the most important prognostic indicator, and end-stage renal disease may occur in up to 5% of patients with nephritis. Long-term follow-up care is indicated in patients with kidney involvement. Recurrent flares of HSP may occur in up to 3% of patients, and in one study had a lag period of 2 to 26 months after the initial presentation.
Supportive care is sufficient for the majority of patients, and nonsteroidal anti-inflammatory agents are useful for significant joint pain. The goals of HSP therapy are to minimize symptoms, decrease short-term morbidities, and prevent chronic renal insufficiency. Systemic corticosteroids have been advocated for patients with severe gastrointestinal, joint, or scrotal involvement, as well as for renal involvement. They are generally not recommended for rash, mild joint pain, or mild abdominal discomfort alone. Various regimens have been recommended for HSP nephritis, including oral methylprednisolone or prednisone, high-dose intravenous pulse methylprednisolone, urokinase, cyclophosphamide, mycophenolate mofetil, azathioprine, plasma exchange, dipyridamole, and heparin/warfarin. The benefits of systemic steroid therapy for HSP remain somewhat controversial. A meta-analysis revealed that corticosteroids reduced the mean resolution time of abdominal pain, reduced the odds of developing persistent kidney disease, and reduced the odds of both surgical intervention and recurrence. However, in a long-term study assessing outcomes 8 years after a randomized placebo-controlled prednisone study in a cohort of 171 HSP patients, there were no differences between prednisone and placebo groups with regard to hematuria, proteinuria, or hypertension. A retrospective cohort study of hospitalized children with HSP revealed an association between decreased hazard ratios for abdominal surgery, endoscopy and abdominal imaging, and early corticosteroid exposure. Taken together, these and other reports suggest the need for more controlled studies of the effect of corticosteroids on HSP outcomes.
Acute Hemorrhagic Edema of Infancy
Acute hemorrhagic edema (AHE) of infancy (AHE of childhood, Finkelstein disease) is a form of leukocytoclastic vasculitis characterized by fever, large purpuric skin lesions, and tender edema and reported most often in infants and children between the ages of 4 months and 3 years. The cutaneous lesions often have a “cockade” (medallion-like) pattern, with scalloped borders and central clearing ( Fig. 21-9 ). They begin as edematous papules with petechiae and expand centrifugally with coalescence to result in the characteristic clinical pattern ( Figs. 21-10 and 21-11 ). Facial edema is common. Although the cutaneous eruption may be impressive, patients are usually otherwise well, and involvement of the gastrointestinal tract, kidneys, and joints is uncommon. Intussusception has been rarely observed. The etiology is unclear, although an infectious trigger (i.e., upper respiratory infection, conjunctivitis, pharyngitis, otitis media, or pneumonia) is hypothesized. Prodromal symptoms most often are limited to those of respiratory tract illness or diarrhea.
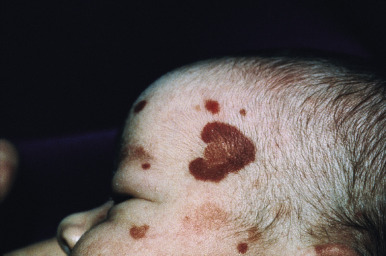
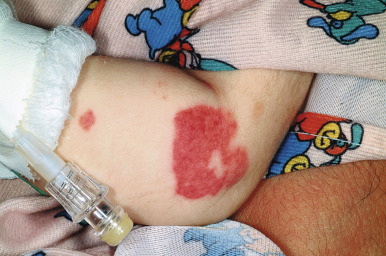
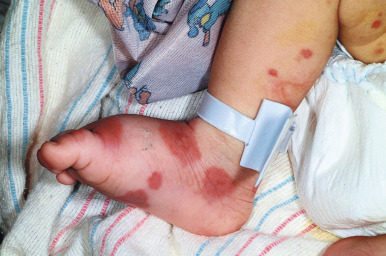
Skin biopsy in AHE reveals leukocytoclastic vasculitis similar to HSP. Direct immunofluorescence studies, however, do not consistently reveal IgA deposition and in up to three-quarters of patients are entirely negative. Laboratory findings may include leukocytosis and an elevated erythrocyte sedimentation rate, but hematuria, proteinuria, and hematochezia are usually absent. The course of AHE is marked by a rapid onset with a short benign course followed by complete recovery, usually over 1 to 3 weeks. No treatment is generally necessary, because AHE is typically a self-limited process. Although the disorder resembles HSP, controversy exists over whether it represents an infantile form of HSP or whether it is a distinct and unrelated clinical entity. Arguments in favor of the latter include the lack of internal organ involvement, the absence of IgA immune deposits in most patients, and the benign course without a propensity toward recurrences.
Hypersensitivity Vasculitis
Hypersensitivity vasculitis ( HV ) (cutaneous small-vessel vasculitis, cutaneous leukocytoclastic vasculitis, cutaneous leukocytoclastic angiitis) is a term used to denote a leukocytoclastic vasculitis involving primarily the skin and in most cases is secondary to a drug ingestion or infectious process. It is most common in adults and significantly less common in children, in whom HSP is the most common form of cutaneous vasculitis. Potential infectious etiologies of HV include Streptococci, hepatitis B and C, nontyphoidal Salmonella, and Mycobacteria (both tuberculous and nontuberculous).
The diagnosis of HV is one of exclusion, and other causes of primary cutaneous vasculitis (i.e., HSP, cryoglobulinemic vasculitis [CV]) or secondary cutaneous vasculitis (i.e., urticarial vasculitis [UV], connective tissue disorders, malignancy, endocarditis, Behçet disease) must be ruled out. The lesions of HV tend to occur in “crops” (groups of lesions of similar age) because of simultaneous exposure to the inciting antigen.
Patients with HV most often present with palpable purpura ( Fig. 21-12 ) and less often with urticaria, vesicles, pustules, superficial ulcers, or necrotic lesions. The lesions may be smaller and petechial in appearance in some patients ( Fig. 21-13 ). Skin involvement is most notable in dependent areas and in areas of trauma or tight-fitting clothing. Symptoms are usually absent, although pain or burning may be present, and systemic involvement is uncommon.
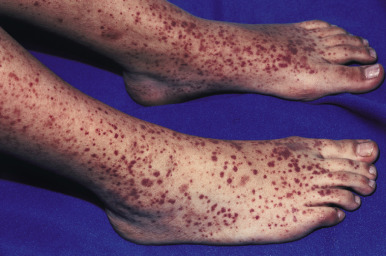
The list of potential drug causes of HV is extensive and includes multiple classes of antibiotics, nonsteroidal anti-inflammatory agents, antiepileptic drugs, insulin, propylthiouracil, omeprazole, and oral contraceptives. (For a more complete listing of potential drug etiologies, see Chapter 20 .) Identification and withdrawal of the offending agent is vital in patients with HV, and in those with skin-limited disease no other specific therapy is usually necessary. For patients with either severe cutaneous or systemic involvement, therapeutic options include corticosteroids, colchicine, dapsone, or other immunosuppressive agents. The lesions of HV usually resolve over weeks to months with resultant postinflammatory hyperpigmentation.
Urticarial Vasculitis
Urticarial vasculitis ( UV ) refers to a type of cutaneous leukocytoclastic vasculitis that presents with urticarial features and is often associated with an underlying systemic disease. In contrast to common urticaria, the lesions of UV are distinct in that they last for longer than 24 to 48 hours, often have a dusky or purpuric appearance, are associated more often with burning than pruritus, and leave postinflammatory hyperpigmentation after resolution ( Fig. 21-14 ). Confirmation of the diagnosis of UV often requires skin biopsy given the potential overlap with other conditions, especially allergic urticaria. Deposits of Igs, complement, or fibrin may be found around blood vessels in UV in up to 80% of patients.
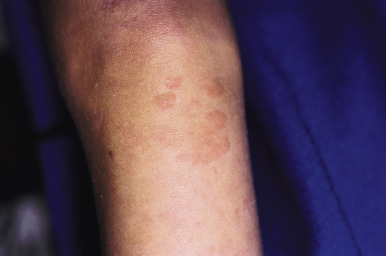
UV may be idiopathic or associated with other diseases including autoimmune disorders, infections, drug reactions, or paraneoplastic syndromes. Most patients can be characterized as having either normocomplementemic UV (in which case the majority represent idiopathic UV) or hypocomplementemic UV (see the following section). Patients with hypocomplementemic UV have a propensity toward more severe multiorgan involvement and systemic disease associations. The most commonly associated autoimmune disorders are systemic lupus erythematosus (SLE) and Sjögren syndrome (see Chapter 22 ). Serum sickness-like reactions to drugs, cryoglobulinemia, and hepatitis C virus (HCV) infection are also associated in some patients with UV.
Hypocomplementemic Urticarial Vasculitis Syndrome
Hypocomplementemic urticarial vasculitis syndrome ( HUVS ) is the terminology used to describe patients with UV in association with angioedema and systemic symptoms, most often obstructive pulmonary disease and ocular inflammation (conjunctivitis, iritis, episcleritis, uveitis). Glomerulonephritis and arthritis may also be present, and the condition is relatively rare in pediatric patients. Decreased serum Clq levels are seen in most patients, and autoantibodies against this factor are detectable in the serum. Although antibodies to Clq are also found in some patients with SLE, HUVS appears to be a unique entity.
The cutaneous lesions of UV are urticarial in nature, favor the trunk and proximal extremities, and often have a purpuric quality. The primary distinguishing features are the persistence of lesions longer than is typical for allergic urticaria (2 to 6 hours), pain or burning, and associated hyperpigmentation as the lesions resolve. If the diagnosis of UV is confirmed, a thorough physical examination and laboratory investigation to assess for associated conditions are indicated. If an underlying disease is identified, specific therapy for that disorder is indicated. There is otherwise no specific therapy for UV. Various therapeutic modalities including prednisone, hydroxychloroquine, indomethacin, azathioprine, colchicine, methotrexate, cyclophosphamide, cyclosporine, intravenous Ig and dapsone have been used but with an inconsistent response.
Cryoglobulinemic Vasculitis
Cryoglobulins are circulating Igs that precipitate at temperatures below 37° C and may be associated with any of several infectious, autoimmune, or malignant disorders. When these immune complexes deposit in blood-vessel walls and activate complement, CV results. Cryoglobulins have been classified into three types based on the presence or absence of monoclonality and their association with rheumatoid factor. Type I (monoclonal cryoglobulinemia) is a monoclonal antibody and is usually associated with a hematologic malignancy, whereas types II and III are composed of a mixed antibody response and hence are termed mixed cryoglobulinemia . The terminology essential mixed cryoglobulinemia is used to describe mixed cryoglobulinemia in the absence of other identified infectious, immunoblastic, or neoplastic disorders. The most common infectious disease to be associated with CV is HCV, which is associated with mixed cryoglobulins. In one large series of adults with SLE, cryoglobulins were detected in the sera of 25% of the patients and were associated with an increased incidence of HCV infection. Patients with CV may experience a variety of skin manifestations including purpura, papules, nodules, skin necrosis, urticaria, livedo reticularis, and bullous or ulcerated lesions. The classic triad of CV is purpura, weakness, and arthralgias, along with possible multiorgan involvement. Treatment for CV should be directed toward the underlying systemic disease when one is identified. The main goals of therapy for mixed cryoglobulinemia are to eradicate HCV infection, delete the underlying B-cell clonal expansions that occur, and deplete the cryoproteins. A variety of anti-inflammatory, antiviral, and immunosuppressive agents (including pegylated interferon-α, ribavirin, and rituximab) have been used to this end with variable success.
Necrotizing Vasculitis with Granulomas
There are several systemic diseases in which necrotizing vasculitis is seen in association with granulomatous changes, including granulomatosis with polyangiitis (GPA), allergic granulomatosis (Churg–Strauss syndrome), and lymphomatoid granulomatosis. The first two will be discussed in this section. These disorders are primarily seen in adults but may occasionally occur in children. They are often associated with antineutrophil cytoplasmic antibodies (ANCAs), which may play a pathogenic role and more importantly are useful in the diagnosis and management of (mainly small-vessel) vasculitides. GPA and Churg–Strauss syndrome, along with the entity microscopic polyangiitis (MPA), have been collectively referred to as ANCA-associated vasculitides. MPA, which is not discussed in detail here, is characterized by a necrotizing small-vessel vasculitis without granuloma formation and with a similar presentation to GPA with less common involvement of the ear, nose, and throat, and occasional limitation to the kidneys. Pulmonary capillaritis and necrotizing glomerulonephritis are seen, as are constitutional symptoms such as fever, malaise, arthralgias, myalgias, and weight loss. Severe anemia may occasionally be the presenting symptom of MPA.
Granulomatosis with Polyangiitis
GPA (formerly known as Wegener granulomatosis ) is a necrotizing granulomatous vasculitis of small- to medium-sized arteries involving the upper and lower respiratory tract in association with kidney involvement and variable degrees of vasculitis in other organ systems. Although it is quite rare, among the primary systemic vasculitides in children GPA is one of the most common and has an annual incidence of 0.03 to 3.2 per 100,000 children. Classification criteria for GPA have been proposed by the ACR and the EULAR/PRES. The latter diagnostic criteria have been demonstrated more sensitive in classifying pediatric GPA than the ACR criteria.
GPA usually presents with constitutional symptoms (malaise, fatigue, fever, and weight loss), and symptoms referable to the upper and/or lower respiratory tracts include cough, rhinorrhea, nasal stuffiness, nasal mucosal erosions or ulcers, earache, or sinusitis. Hemoptysis or pleurisy may be present, and nodular cavitary lesions in the lungs are a characteristic finding on radiographic studies. The nose, nasal sinuses, nasopharynx, glottis, trachea, bronchi, and lungs may all be affected by the necrotizing vasculitis, and subglottic stenosis and nasal deformity may result, the former occurring more commonly in children. Conjunctival injection may be present, as may arthralgias, myalgias, and glomerulonephritis, although pediatric disease may present without renal involvement. End-stage renal failure may occasionally result.
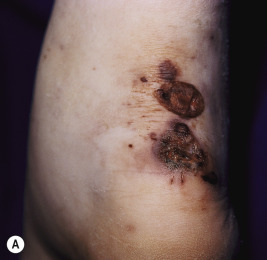
Skin involvement occurs in up to 53% of pediatric patients with GPA. The manifestations are variable but most commonly consist of palpable purpura. Necrotic papules and plaques ( Fig. 21-15 ), subcutaneous nodules, vesicles, pustules, and ulcers resembling pyoderma gangrenosum may occur. Papulonecrotic lesions may favor the extremities (especially the elbows) but may also occur on the face and scalp. Purpura occurring on the lower extremities in conjunction with abdominal pain and hematuria may be the presenting feature of GPA, and this constellation may mimic HSP in the early stage. Gingivitis can occur and presents as spongy, friable, and exuberant tissue with petechiae and preferential involvement of the interdental papillae. Ophthalmologic findings (which may occur in up to 50% of patients) include not only conjunctivitis, but also dacryocystitis, episcleritis, corneoscleral ulceration, retinal artery thrombosis, uveitis, proptosis, cavernous sinus thrombosis, and pseudotumor of the orbits. Pericarditis, endocarditis, and coronary vasculitis have been reported, and peripheral neuropathy and cerebral vasculitis, with or without subarachnoid or intracerebral hemorrhage, have been noted in up to 25% of patients.
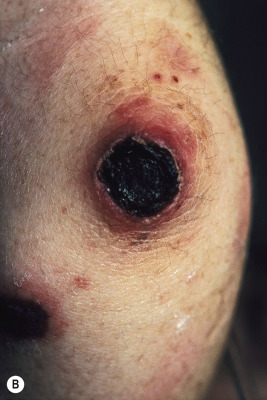
The diagnosis of GPA is suggested by the clinical presentation and confirmed by tissue biopsy examination in conjunction with laboratory findings. Histologic evaluation of tissues (especially skin, lung, or kidney) reveals the characteristic necrotizing granulomatous vasculitis picture, which is highly suggestive of (but not pathognomonic for) GPA within the appropriate clinical context. Up to 80% of patients with GPA have positive cytoplasmic ANCAs (c-ANCAs) and usually negative perinuclear ANCAs (p-ANCAs) on laboratory testing. In one series of pediatric patients with GPA, c-ANCA positivity correlated with kidney involvement.
Before the advent of cytotoxic agents, GPA was nearly always a fatal disease with a mean survival of 5 months. The most commonly used treatment is a combination of immunosuppressive agents, most often glucocorticoids and cyclophosphamide and occasionally azathioprine. Less commonly utilized therapies include plasmapheresis, extracorporeal membrane oxygenation, intravenous immunoglobulin (IVIG), mycophenolate mofetil, and rituximab. Survival rates have improved with the addition of cytotoxic agents to the corticosteroids, but side effects and toxicities related to these drugs are a major source of morbidity. The use of methotrexate is advocated by some, and this agent has shown promise both in induction therapy in combination with glucocorticoids and in maintaining remission of the disease after induction with a cyclophosphamide and glucocorticoid regimen. Cotrimoxazole is often used for treatment of GPA and may have benefit both in the prevention of opportunistic infection and in modifying disease activity. Long-term morbidities related to GPA and its therapy may include infertility, skeletal complications (osteoporosis, avascular necrosis), renal failure, subglottic stenosis, hearing impairment, and nasal septal or upper airway deformities.
Churg–Strauss Syndrome
Churg–Strauss syndrome (CSS, allergic granulomatosis, eosinophilic GPA) is a rare condition characterized by atopic manifestations and a granulomatous vasculitis involving small- and medium-sized arteries. It occurs primarily in males between 30 and 40 years of age and only rarely in children. A distinguishing feature of CSS-associated asthma compared with typical allergic asthma is the late onset in the former, at a mean age of 35 years.
Patients with CSS often experience a prodromal phase during which time they have allergic rhinitis and asthma, and occasionally sinusitis and nasal polyps. After several years, they develop peripheral eosinophilia, eosinophilic tissue infiltration, worsening of their asthma, gastroenteritis, and diffuse pulmonary infiltrates (second phase). The third, or vasculitic, phase presents with fever, weight loss, and widespread vasculitis and inflammation. If left untreated, this phase may result in death. Clinical manifestations may include arthralgias, myositis, peripheral neuropathy, eosinophilic pneumonitis (Löffler syndrome), and occasional renal, cardiac, CNS, or ocular involvement. Cardiac involvement occurs in roughly half of the patients and may include pericarditis, myocarditis, and tamponade. Skin involvement may occur in up to 70% of patients and includes palpable purpura, petechiae, subcutaneous nodules, urticaria, livedo reticularis, and papulonecrotic lesions similar to GPA. Digital ischemic ulcers and Raynaud phenomenon have also been noted.
The ACR criteria for the diagnosis of CSS support the diagnosis when four of the following six criteria are met: asthma, eosinophilia greater than 10%, mononeuropathy/polyneuropathy, nonfixed pulmonary infiltrates, paranasal sinus abnormality, and biopsy containing a blood vessel with extravascular eosinophils. Laboratory studies may be useful, because approximately 70% of adult patients with CSS have a positive ANCAs, usually perinuclear with antimyeloperoxidase specificity (MPO-ANCA). However, in contrast to adult patients, pediatric patients with CSS may be negative for ANCA. In adults, ANCA positivity appears to portend more common ear-nose-throat manifestations (rhinitis, sinusitis, polyps), peripheral nerve involvement, and renal disease, whereas ANCA negativity may predict more common cardiomyopathy.
CSS is treated with glucocorticoids with or without the addition of other immunosuppressive agents (i.e., cyclophosphamide, azathioprine), and the prognosis has traditionally been fairly favorable with a lower mortality rate in comparison to other systemic vasculitides. Combination therapy with other agents including cyclophosphamide, azathioprine, infliximab, or methotrexate may be useful in some patients. Steroid-dependent asthma, however, is common and may persist even after the vasculitis is in remission. In a large series of children with CSS, more common cardiopulmonary manifestations, less common peripheral neuropathy, and higher mortality were noted when compared with the clinical experience in adults. However, in another smaller series, mononeuritis multiplex was present in 60% of children with CSS.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


