Abstract
In this chapter the term neoplasm is used synonymously with tumor in a general, albeit imperfect, sense, consistent with its common usage: an abnormal proliferation (“new growth”) of cells that appears to be relatively autonomous (i.e. not reactive) . Of note, common infantile hemangiomas are discussed separately (see Ch. 103 ) as are telangiectatic lesions (see Ch. 106 ) and vascular malformations (see Ch. 104 ); however, in some patients, vascular malformations may be associated with a number of the cellular vascular proliferations discussed herein. The chapter also includes a review of vascular lesions that were previously designated as vascular tumors (angiomas) but have since been reclassified as distinctive vascular malformations, e.g. glomuvenous malformation (previously “glomangioma”), verrucous venulocapillary malformation (previously verrucous “hemangioma”). In addition, several entities with established or suspected features of reactive processes that mimic neoplasms are discussed as is Kaposi sarcoma, a virus-associated process of as-yet-uncertain position on the hyperplasia–neoplasia spectrum, but with an associated mortality rate. Two benign vascular proliferations of more seemingly straightforward infectious etiology (bacillary angiomatosis and verruga peruana) are covered elsewhere (see Ch. 74 ). With these caveats in mind, a broad working classification of vascular neoplasms and neoplastic-like conditions is presented.
Keywords
vascular tumors, vascular malformations, hemangioma, angioendotheliomatosis, angiokeratoma, hemangioendothelioma, angiosarcoma, Kasabach–Merritt phenomenon, Kaposi sarcoma, glomus tumor, hemangiopericytoma, intravascular papillary endothelial hyperplasia, reactive angioendotheliomatosis, intravascular reactive angioendotheliomatosis, diffuse dermal angiomatosi, hobnail lymphatic malformation, hobnail hemangioma, targetoid hemosiderotic hemangioma, cherry angioma, sinusoidal hemangioma, tufted angioma, multifocal lymphangioendotheliomatosis with thrombocytopenia, glomeruloid hemangioma, microvenular hemangioma, epithelioid hemangioma, angiolymphoid hyperplasia with eosinophilia, pyogenic granuloma, lobular capillary hemangioma, spindle cell hemangioma, kaposiform hemangioendothelioma, Dabska-type hemangioendothelioma, papillary intralymphatic angioendothelioma, retiform hemangioendothelioma, glomuvenous malformation, glomangiosarcoma
Introduction
In this chapter we use the term neoplasm synonymously with tumor in a general, albeit imperfect , sense, consistent with its common usage: an abnormal proliferation (“new growth”) of cells that appears to be relatively autonomous (i.e. not reactive) . Of note, common infantile hemangiomas are discussed separately (see Ch. 103 ) as are telangiectatic lesions (see Ch. 106 ) and vascular malformations (see Ch. 104 ); however, in some patients, vascular malformations may be associated with a number of the cellular vascular proliferations discussed herein. The chapter also includes a review of vascular lesions that were previously designated as vascular tumors (angiomas) but have since been reclassified as distinctive vascular malformations, e.g. glomuvenous malformation (previously “glomangioma”), verrucous venulocapillary malformation (previously verrucous “hemangioma”). In addition, several entities with established or suspected features of reactive processes that mimic neoplasms are discussed as is Kaposi sarcoma, a virus-associated process of as-yet-uncertain position on the hyperplasia–neoplasia spectrum, but with an associated mortality rate. Two benign vascular proliferations of more seemingly straightforward infectious etiology (bacillary angiomatosis and verruga peruana) are covered elsewhere (see Ch. 74 ). With these caveats in mind, a broad working classification of vascular neoplasms and neoplastic-like conditions is presented in Table 114.1 . Selected vascular neoplasms and neoplastic-like proliferations with their associated syndromes and characteristic laboratory findings are outlined in Fig. 114.1 .
| A WORKING CLASSIFICATION OF CUTANEOUS VASCULAR ANOMALIES |
| Reactive conditions |
|
| Telangiectasias (see Ch. 106 ) |
|
| Vascular malformations (see Table 104.2 , Table 104.5 for associated mutated genes ) |
|
| Benign vascular neoplasms and neoplastic-like proliferations |
|
| Borderline and low-grade malignant vascular neoplasms |
|
| Malignant vascular neoplasms |
|
| Perivascular neoplasms and neoplastic-like proliferations |
|
# Not clear if a multifocal vascular neoplasm or malformation.
* Also viewed as a reactive condition.
** Likely a sinusoidal pattern of intravascular endothelial hyperplasia within a pre-existing low-flow vascular malformation.
*** Also viewed as a vascular malformation.
† Reclassified by the WHO as a malignant, rather than intermediate or borderline, vascular tumor.
†† A spectrum of mesenchymal tumors that includes clear cell “sugar” tumor of the lung, angiomyolipoma, lymphangiomyomatosis, clear cell myomelanocytic tumor, and the rare cutaneous tumors with a similar morphology and phenotype.
Benign Vascular Neoplasms and Reactive Hyperplasias
Intravascular Papillary Endothelial Hyperplasia (PEH)
▪ Masson’s pseudoangiosarcoma ▪ Masson’s tumor
- ▪
Not a specific disease entity, but rather a distinct histopathological pattern that can be confused with angiosarcoma
- ▪
The primary or “pure” form is not associated with a pre-existing vascular anomaly and appears as a solitary, slowly growing, often painful nodule located within a dilated dermal, subcutaneous, or submucosal vein
- ▪
Secondary forms are associated with venous malformations and other vascular anomalies prone to thrombosis
- ▪
Rare forms are extravascular, probably arising within hematomas
- ▪
Thought to represent an unusual, exuberant response of endothelial cells to organizing thrombus
Introduction
Masson first described this process in 1923 in hemorrhoidal veins, terming it hemangio-endotheliome vegetant intravasculaire , and interpreted it as a neoplastic process mimicking angiosarcoma. In 1932, Henschen re-interpreted the process as reactive, and in 1971 Kauffman and Stout noted its occurrence not only in thrombosed vessels but also in a hematoma, further demonstrating the potential for confusion with soft tissue angiosarcoma. A lymphatic vessel counterpart of Masson’s lesion was reported in 1979 in a cystic lymphatic malformation .
Epidemiology
Papillary endothelial hyperplasia (PEH) shows a slight female predominance, most pronounced for the rare extravascular examples . Although lesions can occur at any age, most are in adults, with an average age of 34 years. A history of trauma is elicited in a small minority of patients .
Pathogenesis
Areas of papillary endothelial hyperplasia can, in most cases, be seen to merge with definitive thrombus material, in support of the notion that they represent an unusual form of thrombus organization.
Clinical features
Primary lesions of superficial tissues appear as solitary, firm masses, often with red or blue discoloration of the overlying skin or mucosa. The history is typically one of slow growth over a period of several months or years. These occur most commonly within veins of the head and neck and, interestingly, the fingers, but they may also arise elsewhere . In a study of 314 cases, 56% were of the primary form, 40% were associated with other vascular lesions, and 4% appeared extravascularly . Clinical features of the secondary forms are those of the underlying vascular anomaly.
Pathology
Intravascular examples may be limited to the confines of a single thin-walled vein or may arise, often multifocally, within pre-existing vascular lesions such as venous malformations, glomuvenous malformations, spindle cell hemangiomas, and pyogenic granulomas ( Fig. 114.2 ). Foci of intravascular PEH are extremely common in venous malformations and serve to distinguish these low-flow lesions from high-flow arteriovenous malformations. Rarely, the involved vessel wall is ruptured, allowing the proliferative vascular process to spill out into the adjacent stroma. Extravascular examples that on serial sectioning show no evidence of a surrounding blood vessel wall may have arisen within an organizing hematoma .
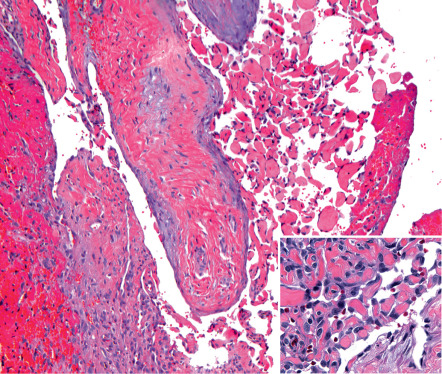
Early lesions show growth of endothelial sprouts into fibrinous thrombus material, dividing it into papillary fronds lined by a single layer of plump endothelial cells with minimal mitotic activity and no significant cytologic atypia ( Fig. 114.2 , inset). The early fibrin cores of the papillae become collagenized and hyalinized with time, and the endothelial lining becomes thin and attenuated. Lesional papillae may fuse to form an anastomosing meshwork of vessels separated by connective tissue stroma that mimics angiosarcoma. However, the relatively high mitotic rate, striking pleomorphism, and necrosis that characterize angiosarcoma are lacking .
Differential diagnosis
The clinical appearance of these lesions is nonspecific, and diagnosis relies on microscopic examination. The most important consideration for the pathologist is well-differentiated angiosarcoma.
Treatment
Surgical excision is usually curative. Local recurrences may occur when the lesion is superimposed on a vascular malformation that may generate new foci of endothelial hyperplasia.
Reactive Angioendotheliomatosis
- ▪
Rare, self-limited process occurring exclusively in the skin and characterized histologically by a dense proliferation of small capillaries
- ▪
The most common form is predominantly intravascular, resulting in luminal obliteration of pre-existing dermal vessels; many of these patients have vaso-occlusive disorders or systemic infections
- ▪
Should be distinguished from intravascular large cell lymphoma (B or NK/T) , an angiotropic lymphoma erroneously termed malignant angioendotheliomatosis prior to the advent of discriminating immunohistochemistry
Introduction
Angioendotheliomatosis has historically been considered to be a single disease entity, divided into benign and malignant variants. As such, the large pleomorphic cells found within blood vessels in malignant angioendotheliomatosis were thought to be transformed endothelial cells. However, immunohistochemical studies have convincingly demonstrated that these cells are neoplastic lymphocytes. Accordingly, malignant angioendotheliomatosis has been renamed intravascular large cell lymphoma (B or NK/T) (see Ch. 119 ). The benign form is a true angioendotheliomatosis and is considered reactive.
Epidemiology
Reactive angioendotheliomatosis is rare and although it can occur at any age, the age of onset often reflects that of any associated systemic disorder. While many cases are idiopathic, this vascular proliferation is associated with systemic disorders such as bacterial endocarditis, monoclonal gammopathies (including type I cryoglobulinemia), antiphospholipid syndrome, renal transplantation, hepatic disease, and rheumatoid arthritis. The diffuse dermal angiomatosis variant occurs most commonly on the leg or the breast and is associated with atherosclerosis, cigarette smoking, and large pendulous breasts.
Pathogenesis
Reactive angioendotheliomatosis is a benign self-limited process. Its frequent association with various systemic conditions has led some investigators to propose that the capillary proliferation may be caused by a circulating angiogenic factor . However, the fact that many of the disorders associated with this process cause vascular occlusion or tissue ischemia suggests that a local, hypoxia-induced increase in vascular endothelial growth factor (VEGF) may be playing a role. In addition, immunological factors may contribute to the proliferative process.
Clinical features
Although erythematous nodules or plaques, often with superimposed petechiae or ecchymoses, are the characteristic primary lesions, the spectrum ranges from erythematous macules to tumor-like masses. Sites of involvement can vary, but the most common is the lower extremities. Focal ulceration may be evident, especially in the diffuse dermal angiomatosis variant (see Fig. 105.20 ).
Pathology
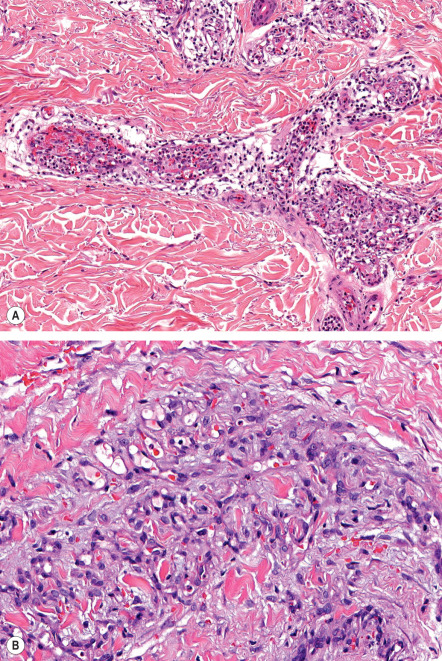
In this predominantly dermal, but also occasionally subcutaneous, process, there is a poorly marginated proliferation of closely packed capillaries lined by plump endothelial cells rimmed by small numbers of pericytes. The pattern of proliferation is highly variable both within and between lesions and is typically described as diffuse and/or lobular . Focally, more dilated capillaries may be present and foci of hemosiderin deposition, fibrin microthrombi, and/or mild chronic inflammation are relatively common. Immunohistochemical positivity for endothelial markers such as CD31 and von Willebrand factor, and focally for pericyte/smooth muscle-associated actins, confirms the cellular composition. Cytologic atypia is absent and mitotic figures are rare to absent.
The capillary lumina are small and may be occluded by fibrin thrombi and bulging endothelial cells ( Fig. 114.3A ). In many cases, the proliferating capillaries appear to be contained within a larger, pre-existing vessel, obliterating its lumen, but sometimes the proliferation extends directly between collagen bundles ( Fig. 114.3B ). Cases associated with cryoglobulinemia typically show intraluminal and intracellular eosinophilic globules, but this finding can also be seen in the absence of cryoglobulinemia.
Differential diagnosis
The clinical differential diagnosis includes vascular tumors (e.g. Kaposi sarcoma, angiosarcoma) as well as other causes of ulceration (see Fig. 105.1 ). Histologically, one must consider other forms of intravascular endothelial proliferation, including intravascular pyogenic granuloma, papillary intralymphatic angioendothelioma, and intravascular PEH. Intralymphatic histiocytosis can be distinguished by the presence of CD68-positive histiocytes within dilated lymphatic vessels.
Treatment
Evaluation for known associated systemic disorders is important because lesions may regress upon resolution of the underlying condition, e.g. revascularization of an ischemic limb. In case reports, oral isotretinoin has led to improvement, possibly due to its antiangiogenic properties .
Angiokeratomas
- ▪
Small, dark, vascular, and variably keratotic lesions that result from dilation of superficial vessels
- ▪
Angiokeratoma circumscriptum is a capillary–lymphatic or capillary malformation
- ▪
Angiokeratoma corporis diffusum results from several lysosomal storage disorders and is associated with systemic manifestations
Introduction
Angiokeratomas are well-circumscribed vascular lesions consisting of superficial vascular ectasia and hyperkeratosis. Five variants have been recognized. With the exception of angiokeratoma circumscriptum (which represents a capillary–lymphatic or capillary malformation), angiokeratomas result from ectatic dilation of pre-existing vessels in the papillary dermis.
Clinical features
Solitary or multiple angiokeratomas
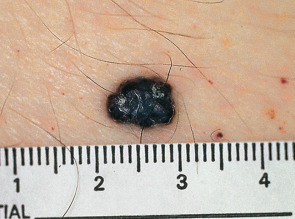
Most commonly, they present as a small, warty, black papule on the lower extremities, but may occur anywhere on the body ( Fig. 114.4 ). The lesions are thought to result from injury to or chronic irritation of the wall of a venule in the papillary dermis. Solitary lesions may be confused with melanoma due to their dark color. Dermoscopy will readily distinguish between these two entities (see Ch. 0 ).
Angiokeratomas of the scrotum and vulva
▪ Angiokeratoma of Fordyce
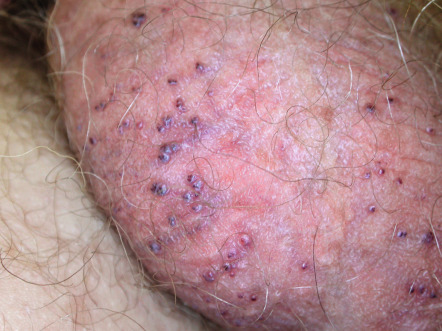
Angiokeratoma corporis diffusum
This disorder is characterized by the development of multiple, often clustered angiokeratomas, usually in a bathing trunk distribution. Lesions vary in number (from only a few to numerous) and usually begin to appear during late childhood or adolescence. X-linked recessive Fabry disease is the best-known entity with this clinical presentation and results from a deficiency of the lysosomal enzyme α-galactosidase A (see Ch. 63 ). This leads to the accumulation of the neutral glycolipid ceramide trihexidose within lysosomes of multiple cell types. Other enzyme deficiencies associated with angiokeratoma corporis diffusum are outlined in Table 63.7 .
Angiokeratoma of Mibelli
Lesions usually develop between the ages of 10 and 15 years and are most commonly situated on the dorsal and lateral aspects of the fingers and toes. They may also occur on the dorsa of the hands and feet and rarely on the elbows and knees. Angiokeratoma of Mibelli may be associated with chilblains and acrocyanosis. In rare instances, ulceration of the fingertips can occur. There is a familial predisposition and the disorder may be transmitted in an autosomal dominant fashion with variable penetrance.
Angiokeratoma circumscriptum
This entity usually develops during infancy or childhood as either a plaque of multiple discrete papules ( Fig. 114.6 ) or hyperkeratotic papules and nodules that often become confluent. They occur on the trunk, arms or legs and are unilateral in most patients. There is a female predominance.
Pathology
Marked dilatation of the papillary dermal vessels is seen in association with an acanthotic, variably hyperkeratotic epidermis. Elongated rete ridges may partially or completely enclose vascular channels, and a collarette may be present at the margin of the lesions. In Fabry disease, vacuoles can be detected within endothelial cells and pericytes. The amount of glycolipid is small and may be difficult to detect in routinely prepared sections. However, the deposits stain positively with PAS and anti-GB3 antibody. They can also be demonstrated by electron microscopy.
Differential diagnosis
Clinically, angiokeratomas should be distinguished from other vascular lesions as well as acral pseudolymphomatous angiokeratomas (see Ch. 121 ). Darkly colored or thrombosed angiokeratomas may resemble cutaneous melanoma.
Treatment
Patients may request removal for cosmetic reasons. This may be achieved by shave excision, diathermy or laser therapy, the choice of which would largely depend on the size of the lesion.
Targetoid Hemosiderotic Lymphatic Malformation (Hobnail “Hemangioma”)
▪ Targetoid hemosiderotic hemangioma ▪ Superficial hemosiderotic lymphovascular malformation ▪ Hobnail lymphatic malformation
- ▪
Uncommon acquired vascular lesion that usually presents as a red–blue or brown papule, most commonly on the extremities and often with a history of local trauma
- ▪
The papule is sometimes surrounded by a pale ring and then an ecchymotic halo
- ▪
Biphasic pattern of dilated, thin-walled vessels with hobnail endothelial cells in the superficial dermis, transitioning to smaller, slit-like vessels in the deeper dermis
- ▪
Lesional endothelial cells strongly express the lymphoendothelial marker podoplanin and have minimal expression of CD34, consistent with lymphatic differentiation
Introduction and history
The original description in 1988 coined the term targetoid hemosiderotic hemangioma to emphasize the targetoid clinical appearance and hemosiderin deposition. Since then it has become apparent that only a minority of these lesions have an ecchymotic halo and hemosiderin deposition is variable. The term hobnail hemangioma was proposed to encompass lesions with the characteristic biphasic growth pattern and marked hobnail endothelial morphology, with or without a targetoid clinical appearance and/or marked hemosiderin deposition . A benign clinical course has been observed in the over 100 cases that have been reported to date . Recently, this “hemangioma” was reclassified as a targetoid hemosiderotic lymphatic malformation (THLM) .
Epidemiology
There is no gender predilection . In a series of 62 patients, their ages ranged from 6 to 72 years (median, 32 years) .
Pathogenesis
This vascular anomaly was thought to represent the benign end of a spectrum of vascular tumors that have hobnail endothelial cells and which includes papillary intralymphatic angioendothelioma (PILA) and retiform hemangioendothelioma (see Table 114.1 ) . However, the endothelial cells of THLM and PILA, but not retiform hemangioendothelioma, stain positively for several markers of lymphatic differentiation . In addition, the endothelial cells of THLM are generally negative for Wilms tumor-1 and have a low Ki-67 proliferation index. The latter explains its reclassification as a vascular malformation based upon International Society for the Study of Vascular Anomalies (ISSVA)-sanctioned restriction of the suffix “angioma” to intrinsically proliferative vascular tumors .
Trauma may stimulate the appearance of a THLM, with microshunts between small lesional blood vessels and adjacent lesional lymphatic vessels explaining the presence of erythrocytes within lymphatic vessels and hemosiderin deposits .
Clinical features
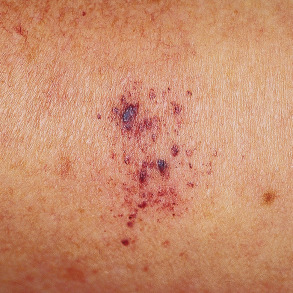
Most lesions are asymptomatic, solitary, well-circumscribed, red–blue to brown papules that are initially 2 to 3 mm in diameter and then slowly increase in size. In some cases, the papule is surrounded by a thin pale ring and then an ecchymotic halo ( Fig. 114.7 ). The ecchymotic ring may fade and eventually disappear over time, and some lesions undergo cycles of spontaneous regression and recurrence. Favored sites, in decreasing order of frequency, are the lower extremities, upper extremities, back, buttock/hip, and chest wall. Lesions have also been reported on the tongue and gingiva .
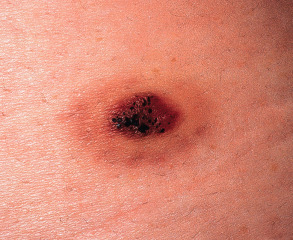
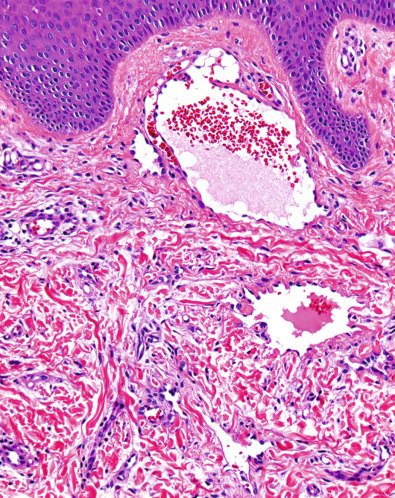
Pathology
There is a biphasic histologic pattern, consisting of: (1) dilated, thin-walled vessels containing small numbers of red blood cells and lined by prominent hobnail endothelial cells within the superficial dermis ( Fig. 114.8 ); and (2) smaller, more slit-like vessels that dissect between collagen bundles in deeper portions of the dermis. Extravasated red blood cells and hemosiderin deposits are common, but may be absent. Although the superficial vessels occasionally have delicate intraluminal papillary fronds, the more complex, multilayered endothelial tufts of PILA are lacking. The endothelial cells of both the dilated and the slit-like vessels stain positively for the lymphoendothelial markers VEGF-3 and podoplanin (see Ch. 102 ), but show little, if any, reactivity for the blood vascular marker CD34 .
Differential diagnosis
The clinical differential diagnosis includes melanocytic nevus, sclerosing hemangioma, and benign vascular neoplasms and proliferations in Table 114.1 . Histopathologically, it must be distinguished from patch-stage and lymphangioma-like variants of Kaposi sarcoma, well-differentiated angiosarcoma, retiform hemangioendothelioma, PILA, and microcystic lymphatic malformation.
Treatment
Treatment consists of simple excision. Longitudinal evaluation of 35 patients, ranging from 1 to 4 years, revealed no local recurrence or systemic metastasis .
Verrucous Venulocapillary Malformation (Verrucous “Hemangioma”)
▪ Verrucous venous malformation
- ▪
Rare congenital vascular anomaly that has been reclassified as a vascular malformation
- ▪
Somatic missense mutations in mitogen-activated protein kinase kinase kinase 3 gene ( MAP3K3 ) detected in some lesions
- ▪
Isolated, grouped, or confluent red-to-purple papules with reactive hyperkeratosis; often darkens with age
- ▪
Most commonly occurs on the distal extremity and can be complicated by ulceration and bleeding but not local tissue hypertrophy
- ▪
Histologically, dilated capillaries and venules are present within the papillary and investing dermis and subcutis but not the reticular dermis
Introduction and history
This rare, but increasingly well-recognized, vascular anomaly was originally thought to be a “hemangioma”, but has since been reclassified as a vascular malformation. The debate arose in part because these lesions may show evidence of cellular proliferation during their evolution . Mulliken and colleagues recommended use of the term verrucous venous malformation while others prefer the designation verrucous venulocapillary malformation, congruent with the predominant component vessels.
Epidemiology
There are no gender, racial or ethnic predilections.
Pathogenesis
Somatic missense mutations in the mitogen-activated protein kinase kinase kinase 3 gene ( MAP3K3 ) were detected in 6 of 10 verrucous venulocapillary malformations . The mutant allele frequencies ranged from 6–19% in affected tissue and these mutations were not found in unaffected tissue or other types of vascular anomalies. Of note, studies in MAP3K3 knockout mice had previously implicated MAP3K3 in vascular development.
Clinical features
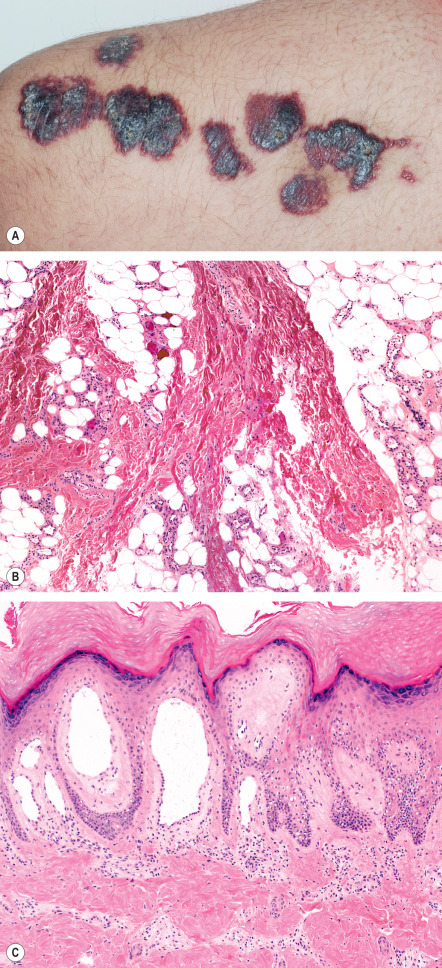
Verrucous venulocapillary malformations are congenital and present as isolated, grouped or confluent red-to-purple papules. The vast majority occur on the distal extremities, particularly the leg ( Fig. 114.9A ) ; the trunk is a rare location . They progressively darken and become hyperkeratotic during childhood, often increasingly complicated by ulceration, bleeding, and scarring. They do not regress and are not associated with local tissue hypertrophy or other developmental anomalies.
Pathology
Features can vary with age, in particular the appearance of secondary orthohyperkeratosis and verrucous hyperplasia, but consistently there are dilated capillaries and venules (without a smooth muscle layer) within the papillary dermis and occasionally the investing dermis around adnexae. There is sparing of the reticular dermis, with the dilated blood vessels reappearing in the subcutis, where they are usually smaller in caliber, often grouped, and sometimes have intraluminal thrombi ( Fig. 114.9B ). The superficial dermal vessels are usually the most obviously dilated with expansion of the dermal papillae ( Fig. 114.9C ).
Endothelial cells consistently express markers of blood vascular differentiation, including CD31 and CD34. Focal positivity for lymphatic endothelial markers, e.g. Prox1, podoplanin, has been observed in some, but not all, studies . Mitotic activity is very low. Many verrucous venulocapillary malformations have light, focal immunoreactivity for GLUT1, in contrast to the intense endothelial GLUT1 positivity seen in infantile hemangiomas ; staining for CD15 and indolamine 2,3 deoxygenase is negative (personal observation).
Differential diagnosis and treatment
The differential diagnosis includes other venous malformations (see Ch. 104 ) and infantile hemangiomas. Therapeutic options include surgical excision and treatment of the superficial component with pulsed dye, long-pulsed Nd:YAG, and/or carbon dioxide lasers.
Pyogenic Granuloma
▪ Lobular capillary hemangioma ▪ Granuloma pyogenicum ▪ Tumor of pregnancy ▪ Eruptive hemangioma ▪ Granulation tissue-type hemangioma
- ▪
Rapidly growing, friable, red papule or polyp of skin or mucosa that frequently ulcerates; most common in children and young adults
- ▪
Those arising on the gingiva of pregnant women ( granuloma gravidarum ) are considered a separate subgroup, but are histologically indistinguishable
- ▪
Consist of lobules of small capillaries set in a fibromyxoid matrix, often distinctly exophytic and bounded by collarettes of hyperplastic epithelium
- ▪
Occasionally found in subcutaneous or intravascular locations
- ▪
Neither infectious in etiology nor granulomatous histologically; often considered a reactive vascular hyperplasia rather than a neoplasm
Introduction and history
Poncet and Dor are credited with the initial description in 1897, and they believed the cause was a Botryomyces infection. The term granuloma pyogenicum was coined in 1904 by Hartzell to describe four similar cases that he thought represented a nonspecific granulation tissue-type response to any type of pyogenic agent . Although the etiology of these common lesions remains uncertain, the term pyogenic granuloma is clearly a misnomer. There is no evidence to implicate any infectious agent, and the histologic appearance is not granulomatous.
Epidemiology
Pyogenic granulomas can develop at any age but are more common in children and young adults. There may be a slight male predominance, but no racial or familial predisposition. Gingival lesions are relatively common during pregnancy (see Ch. 27 ).
Pathogenesis
Pyogenic granulomas exhibit a number of clinical features suggestive of reactive neovascularization, including a common association with a pre-existing injury or irritation, limited capacity for growth, and a propensity for multiple eruptions that may be localized or disseminated. The occasional eruption of pyogenic granulomas within existing port-wine stains and other vascular malformations suggests that abnormalities in blood flow may be etiologically important in some cases.
Clinical features
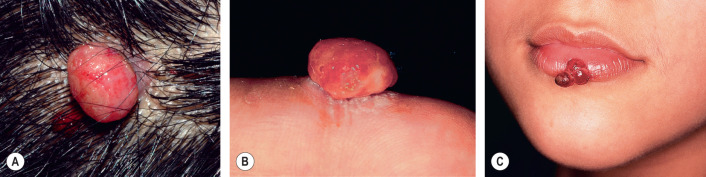
The lesion presents as a solitary red papule or polyp that grows rapidly over the course of several weeks or months, stabilizes, and then may decrease in size ( Fig. 114.10 ). Its final size is rarely >1 cm, and it may persist indefinitely if not removed. Approximately one-third develop following minor trauma. In one series of 289 cases, the most common sites, in decreasing order of frequency, were the gingiva, fingers, lips, face, and tongue . They are extremely friable, frequently ulcerate, and may bleed profusely with minor trauma. Multiple satellite lesions occasionally develop near a primary pyogenic granuloma, usually after destruction of that lesion . Rarely, pyogenic granulomas arise within the subcutis, develop intravascularly, or erupt in a disseminated fashion . Pyogenic granulomas have been reported in association with systemic retinoids , indinavir, and BRAF and EGFR inhibitors, but in some cases this may represent excessive granulation tissue.
Pathology
The quintessential lesion is a well-circumscribed, exophytic, sometimes pedunculated, proliferation of small capillaries, often arranged in a lobular pattern ( Fig. 114.11 ). Lesional capillaries are lined by flattened to slightly plump endothelial cells, rimmed by pericytes, and surrounded by a variably edematous fibromyxoid interstitial stroma containing fibroblasts. Endothelial and stromal cell mitotic activity is highly variable and depends on the stage of growth; a scant infiltrate of lymphocytes, plasma cells, and mast cells may be present. Capillary lumina within the lobules vary from small and angular to branching and ectatic, and foci of thrombosis and intravascular PEH may be present. A few larger vessels with smooth muscle walls, usually venous (but often including a small ascending arterial feeder), are frequently present at the base of the lesion. Thick, intervening bands of dense fibrous tissue sharply define the lobularity and help distinguish pyogenic granuloma from other lobular forms of capillary proliferation such as infantile hemangioma.
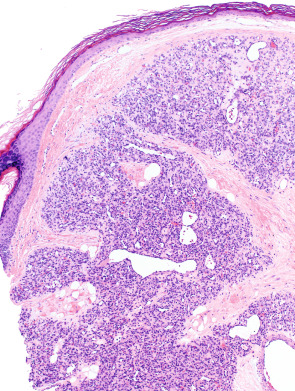
The lateral margins are often defined by prominent epithelial “collarettes” resulting from peripheral adnexal hyperplasia or, in some cases, from downward growth of rete ridges, bridged by flattened epidermis. The histology of many early lesions is altered by ulceration and secondary inflammatory changes, leading to an appearance similar to granulation tissue, with radially oriented capillaries, fibrin deposition, and a loss of lobularity. Intravascular and subcutaneous forms demonstrate features similar to superficial lesions, but without the inflammatory complications. Late-stage lesions are characterized by increased intra lobular and inter lobular fibrosis, as well as quiescent, flattened capillary endothelia.
Differential diagnosis
The diagnosis of pyogenic granuloma can be made clinically if the red, bleeding papule is coupled with the characteristic location and history. Otherwise they can be easily confused with amelanotic melanoma and, in the immunosuppressed patient, bacillary angiomatosis or Kaposi sarcoma. Glomus tumors, hemangiomas, and irritated melanocytic nevi and warts can all mimic pyogenic granulomas. Histologic confirmation is always helpful in cases where the diagnosis is in question.
Treatment
Shave excision followed by electrosurgery of the base under local anesthesia is sufficient for most lesions. Patients and parents should be alerted to the possibility of recurrence after removal. Excision with suturing may result in less postoperative bleeding and a lower recurrence rate.
Pulsed dye laser has also been shown to be a safe and effective treatment for small pyogenic granulomas and may be particularly useful in children . There is a report of successful sclerotherapy in nine patients using monoethanolamine oleate, with inconspicuous scar formation and no recurrence .
Cherry Angioma
▪ Cherry hemangioma ▪ Senile angioma ▪ Campbell–De Morgan spot
- ▪
Bright red, dome-shaped to polypoid papules up to several millimeters in diameter that begin appearing during adult life, most commonly on the trunk and upper extremities
- ▪
A common, benign lesion found in most individuals by the age of 60 years, often in large numbers
- ▪
Consist of dilated, congested capillaries and postcapillary venules within the papillary dermis
Introduction and history
Cherry angiomas are the most common of the acquired cutaneous vascular proliferations. They can be unattractive and rarely when eruptive herald an underlying systemic abnormality.
Epidemiology
Both sexes are affected equally. Although they may occasionally develop during adolescence, cherry hemangiomas usually first appear during the third decade of life or later, and then increase in number over time. Most people over 60 years of age have one or more such lesions.
Pathogenesis
An increased number of cherry angiomas often appear during pregnancy and may involute in the postpartum period, suggesting that hormonal factors may be important in their pathogenesis. Two women with hundreds of eruptive cherry hemangiomas, both of whom had increased serum levels of prolactin, have also been reported . Most angiomas that appear in the setting of POEMS syndrome are cherry angiomas, often in association with glomeruloid hemangiomas.
Clinical features
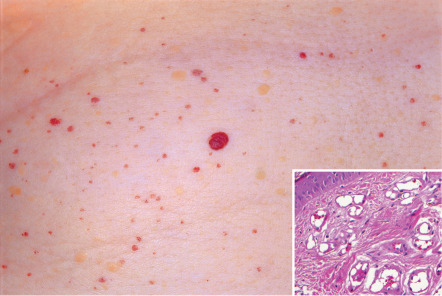
Cherry angiomas are round to oval, bright red to purple, dome-shaped papules ranging in size from barely visible to several millimeters in diameter ( Fig. 114.12 ). Well-developed lesions may be polypoid. They most commonly develop on the trunk and proximal extremities and are rare on the hands, feet, and face. It is not unusual for elderly adults to have 50–100 cherry angiomas on their trunk. Rarely, a segmental distribution pattern, possibly reflecting mosaicism, is seen.
Patients are usually aware of the benign nature of these extremely common lesions and bring them to a physician’s attention only when concerned about their appearance. They are generally asymptomatic, but may occasionally bleed when traumatized.
Pathology
Histologically, congested, ectatic capillaries and postcapillary venules are seen within the papillary dermis and superficial reticular dermis ( Fig. 114.12 , inset). Early lesions are characterized by small lumina and plump endothelial cells. With maturation, the vessels dilate and the endothelial cell cytoplasm flattens, often producing slightly hobnailed nuclei. Vessel diameter decreases with descent into the reticular dermis. There is loss of epidermal ridges centrally and peripherally adnexal epithelial collarettes are seen.
Differential diagnosis
The distinctive clinical and dermoscopic findings (see Ch. 0 ) limit other diagnostic considerations. Glomeruloid hemangiomas, although similar in clinical appearance, can be distinguished histologically. Tiny cherry angiomas may resemble petechiae.
Treatment
Patients may request removal of cosmetically undesirable or chronically traumatized cherry angiomas. This can be accomplished by shave excision, electrodesiccation or laser ablation, and recurrences are unusual.
Tufted Angioma
▪ Acquired tufted angioma ▪ Angioblastoma of Nakagawa ▪ Tufted hemangioma ▪ Hypertrophic hemangioma ▪ Progressive capillary hemangioma
- ▪
Usually presents as an acquired lesion in children and young adults, but may be congenital
- ▪
Inhomogeneous pink to red patches and plaques with superimposed papules that spread slowly, often to involve large areas, then stabilize; rarely completely regress
- ▪
Most commonly located on neck or trunk
- ▪
When congenital can be associated with Kasabach–Merritt phenomenon
- ▪
Generally thought to represent a mild superficial form of kaposiform hemangioendothelioma
- ▪
Characterized histologically by tightly packed tufts of tiny capillaries distributed within the dermis and subcutis in a “cannonball” pattern; foci of spindled endothelial cells may also be present
Introduction
Tufted angioma has often been lumped erroneously with infantile hemangioma under the generic term “capillary hemangioma”. However, a distinction is important given its association with Kasabach–Merritt phenomenon in some infants. In older children and adults, it may clinically mimic Kaposi sarcoma. Current opinion is that tufted angioma falls within a spectrum shared by kaposiform hemangioendothelioma (KHE), with the latter usually having deeper involvement of soft tissues.
History
Tufted angioma was first described in 1989 by Wilson-Jones and Orkin . Apparently identical lesions had been described many years previously as angioblastoma and progressive capillary hemangioma . The association of tufted angioma with Kasabach–Merritt phenomenon, shared by KHE, is now widely recognized .
Epidemiology
Most lesions occur in young adults and children, many during the first year of life. Over 50% present before 5 years of age , and ~15% are congenital . Rarely, lesions appear late in life . Although one family with several affected family members has been reported , the vast majority of cases are sporadic.
Pathogenesis
Tufted angioma and KHE have a significant degree of overlap in histologic features, with resection specimens of KHE often demonstrating dermal changes that would have been interpreted in small biopsy specimens as tufted angioma. In light of the virtually exclusive association of these two entities with Kasabach–Merritt phenomenon, this histologic overlap suggests that tufted angioma may represent a more superficial, milder form of KHE (see KHE section for further discussion of pathogenesis) .
Clinical features
Tufted angiomas appear as mottled red patches or plaques with superimposed angiomatous papules, typically on the neck, trunk or shoulders, that grow slowly by lateral extension over a period of 5 months to 10 years ( Fig. 114.13 ). Occasionally, lesions have an associated growth of lanugo hair or port-wine-like stain . Platelet trapping (Kasabach–Merritt phenomenon) may develop in congenital cases, although less commonly than in KHE. Lesions eventually stabilize in size, then may persist unchanged, shrink, or leave a fibrotic residuum. Rarely, complete spontaneous regression has been observed . Occasionally lesions are painful, and exacerbations of pain may occur during periods of uncontrolled platelet trapping .
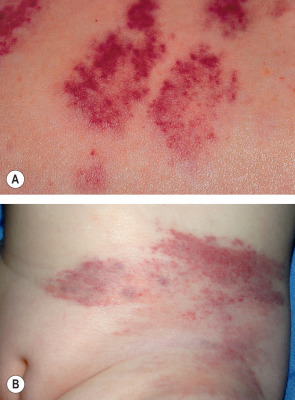
Pathology
Tufted angiomas, as originally described in the dermatology literature, are characterized by multiple discrete lobules of capillaries set within the dermis and often the subcutis in a so-called “cannonball” pattern ( Fig. 114.14 ). Borders of the area of tissue involvement are poorly defined. The dermal collagen and subcutis separating capillary lobules may be histologically normal, but are often fibrotic. The lobules are composed of tiny capillaries with pinpoint lumina, occasionally containing fibrin microthrombi, that are tightly packed together without intervening stroma and frequently bulge into peripherally placed thin-walled vessels ( Fig. 114.14 , inset). Mitotic figures are rare.
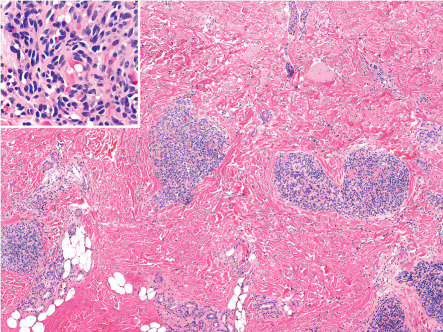
Endothelial cells may be focally spindled, but less prominently than in KHE. The sweeping spindled cell fascicles and epithelioid nodules of KHE are typically absent in tufted angioma . Of note, the endothelial cells of tufted angioma do not immunoreact for infantile hemangioma-associated antigens such as GLUT1 and Lewis Y antigen . Ultrastructural studies have demonstrated classical Weibel–Palade bodies. In cases associated with Kasabach–Merritt phenomenon, platelet trapping has been confirmed with CD61 immunoreaction .
Differential diagnosis
Congenital/early infantile tufted angiomas must be differentiated from infantile hemangiomas, the most common tumors of infancy (see Ch. 103 ). The latter are positive for placental capillary-associated endothelial markers (e.g. GLUT1) and show more rapid growth (without lateral extension) during the first year of life, followed by spontaneous, slow involution . Infantile hemangiomas are transiently positive for the lymphatic-associated marker LYVE-1 early in their course, presumably as an expression of their embryonic, cardinal vein-type endothelial phenotype , but lack other lymphatic markers such as podoplanin and Prox1 .
Other entities that may mimic the clinical appearance of tufted angioma include pyogenic granuloma arising within a vascular malformation and Kaposi sarcoma. Pyogenic granuloma can be distinguished histologically by its characteristic edematous stroma, granulation tissue-type changes, lack of association with lymphatic markers, and generally larger, more loosely packed lesional vessels. Kaposi sarcoma differs in its lack of tufting, presence of a plasma cell infiltrate, and prominence of fascicles of spindle cells forming slit-like spaces. KHE, although overlapping in histology and likely synonymous in pathogenesis, is traditionally described as a more bulky, deeply seated lesion that tends to infiltrate across multiple tissue planes and has more prominent spindled endothelial cells .
Treatment
Complete surgical excision is the treatment of choice for small tufted angiomas, but recurrence is common. Use of the pulsed dye laser has been ineffective , although the argon tunable dye laser with its higher risk of scarring has been used successfully. Both aggravation and improvement have been reported with the Nd:YAG laser. Historically, high-dose systemic corticosteroids were sometimes employed , even though they are generally ineffective. Interferon-α may produce partial regression, but its use has declined given the risk of permanent spastic diplegia in infants (see Ch. 103 ) . For patients with tufted angioma/KHE who develop Kasabach–Merritt phenomenon, first-line therapy consists of vincristine plus prednisone or sirolimus (rapamycin) . Aspirin may also help to control the platelet interaction, pain, and growth of tufted angiomas .
Glomeruloid Hemangioma
- ▪
A histologically distinct type of hemangioma that occurs in patients with POEMS syndrome
- ▪
In POEMS syndrome, presents as multiple, firm, angiomatous papules or plaques scattered primarily on the trunk and proximal extremities
- ▪
Characterized histologically by nests of capillaries within dilated dermal vessels, resembling renal glomeruli
- ▪
May be induced by increased circulating levels of VEGF
Introduction and history
The term glomeruloid hemangioma was coined by Chan et al. in 1990 to describe a distinctive vascular proliferation that occurs in patients with POEMS syndrome ( p olyneuropathy, o rganomegaly, e ndocrinopathy, m onoclonal gammopathy, and s kin lesions). Although the majority of patients with POEMS syndrome have an underlying plasma cell dyscrasia, this lesion occurs more commonly in those with multicentric Castleman disease. There is some overlap with reactive angioendotheliomatosis, but glomeruloid hemangioma is discussed separately because of its highly distinctive histologic features and well-recognized clinical association.
Clinical features
The reported incidences of angiomas in patients with POEMS syndrome range from 25% to 45%. These appear as multiple, firm, dome-shaped, red to purple papules or plaques ranging in diameter from a few millimeters to a few centimeters; they are scattered predominantly over the trunk and proximal extremities (see Ch. 53 ). Additional cutaneous manifestations of POEMS syndrome are outlined in Table 114.2 , as are the types of vascular lesions that occur in these patients. True glomeruloid hemangiomas appear to be fairly specific for POEMS syndrome, and rarely they may appear years prior to recognition of the syndrome . In addition, there are reports of a solitary variant that is not associated with POEMS syndrome.
| POEMS SYNDROME – CLINICAL CRITERIA, CUTANEOUS FINDINGS AND ASSOCIATED VASCULAR LESIONS |
| Diagnostic criteria |
| M onoclonal plasmaproliferative disorder – and – Sensorimotor p olyneuropathy plus at least one other major criterion and one minor criterion |
| Major criteria |
|
| Minor criteria |
|
| Cutaneous findings |
|
| Types of vascular lesions |
|
Pathogenesis
Glomeruloid hemangiomas are thought to be reactive, rather than neoplastic. Patients with POEMS syndrome often have increased circulating levels of VEGF such that this elevation represents a major diagnostic criterion (see Table 114.2 ). Most cases of POEMS syndrome associated with multicentric Castleman disease (~90%) have evidence of human herpesvirus-8 (HHV-8) infection by PCR. It is suspected that viral interleukin (IL)-6 produced by the HHV-8 may indirectly promote angiogenesis by inducing VEGF expression .
Pathology
Most of the angiomas seen in patients with POEMS syndrome are found upon histologic examination to be consistent with common cherry angiomas, with numerous dilated dermal capillaries lined by flattened endothelial cells (see above). Only a few demonstrate the distinctive features of glomeruloid hemangioma. These consist of dilated dermal vessels filled by small, well-formed capillary loops that lead to structures reminiscent of renal glomeruli ( Fig. 114.15 ).
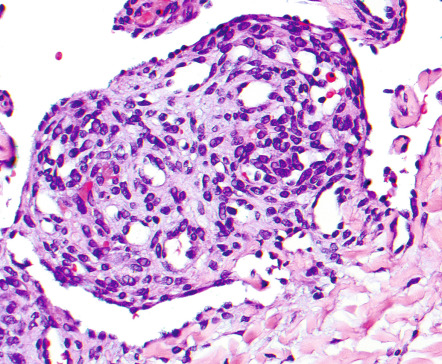
The capillaries comprising these glomeruloid formations are lined by flat endothelial cells surrounded by an outer layer of pericytes and are separated by a scant stroma containing large cells with lightly eosinophilic cytoplasm and multiple eosinophilic globules. The latter are PAS-positive, diastase-resistant, and immunopositive for polytypic immunoglobulin, presumably derived from the associated paraprotein . Of note, these interstitial, immunoglobulin-containing cells are of endothelial derivation. Lastly, since many POEMS syndrome patients have both cherry angiomas and glomeruloid hemangiomas, and the cherry angiomas may display focal glomeruloid formation, it is likely that these two histologic subsets reflect different stages of the same process .
Differential diagnosis
The clinical differential diagnosis includes other vascular proliferations and tumors (see Table 114.1 ) while the histologic differential diagnosis of glomeruloid hemangioma is essentially that described above for reactive angioendotheliomatosis. A more recently described entity, papillary hemangioma , also enters the histologic differential diagnosis . The latter is characterized by intravascular papillary growths of capillaries, pericytes, and stromal cells within ectatic dermal vessels, accompanied by hyaline globules (features reminiscent of glomeruloid hemangioma), but it differs by being solitary, being confined to the head and neck region, and having neither a true glomeruloid architecture nor an association with POEMS syndrome.
Treatment
Treatment is not required, although shave excision, cryosurgery, electrodesiccation, or pulsed dye laser surgery will remove the vascular tumors.
Microvenular Hemangioma
▪ Microcapillary hemangioma
- ▪
An uncommon, acquired, slowly growing, benign vascular tumor of young to middle-aged adults
- ▪
Presents as a small, usually solitary, sharply circumscribed red papulonodule or plaque on the trunk or extremities, with a predilection for the forearms
- ▪
Consists of small branching capillaries and venules with collapsed lumina and conspicuous pericytes infiltrating the full thickness of the reticular dermis
- ▪
May be subject to hormonal influences in some women
Introduction and history
Microvenular hemangioma was first described by that name in 1991 by Hunt, Santa Cruz and Barr , although cases reported as microcapillary hemangiomas are probably identical.
Epidemiology
Reported cases have been in young and middle-aged adults of both sexes. Some cases in women have been temporally related to pregnancy or oral contraceptive use, although this has not been widely corroborated.
Pathogenesis
Microvenular hemangiomas have been noted in patients with POEMS syndrome , suggesting that these lesions, like glomeruloid hemangiomas, may be of reactive etiology. Observations that some microvenular hemangiomas affecting women present in association with pregnancy or oral contraceptive use arguably suggest a hormonal influence.
Clinical features
Microvenular hemangioma typically presents as a solitary, purple to red, slowly enlarging papule, plaque or small nodule on the trunk or extremities (rarely the face). Multiple lesions have occasionally been observed. There appears to be a particular predilection for the forearms, and most lesions are <2 cm in diameter . Although the majority are asymptomatic, mild erythema and tenderness have been reported.
Pathology
Histologically, lesions are poorly circumscribed proliferations of small, relatively monomorphous, branching capillaries and venules involving the full thickness of the reticular dermis. Unlike port-wine stains, in which vascular ectasia is prominent, the vascular lumina of this tumor are inconspicuous and often collapsed. Cells lining the vessels show no atypia and are strongly positive for endothelial markers and are associated with smooth muscle actin-positive pericytes. The vessels are not as delicate or angulated as the lymphatic-like vessels of Kaposi sarcoma or vessels of microcystic lymphatic malformations and often contain red blood cells. The pericytic component is obvious even in routine H&E-stained sections.
Differential diagnosis
The major histologic differential diagnostic consideration for these acquired lesions is patch-stage Kaposi sarcoma. Kaposi sarcoma differs by the presence of delicate lymphatic-like vessels, plasma cells, eosinophilic globules, interstitial fascicles of spindle cells, and positivity for HHV-8.
Treatment
Surgical excision has been effective in reported cases.
Epithelioid Hemangioma
▪ Angiolymphoid hyperplasia with eosinophilia (AHE) ▪ Pseudopyogenic granuloma ▪ Inflammatory angiomatous nodule ▪ Papular angioplasia ▪ Inflammatory arteriovenous hemangioma ▪ Intravenous atypical vascular proliferation ▪ Nodular angioblastic hyperplasia with eosinophilia and lymphofolliculosis ▪ Histiocytoid hemangioma
- ▪
Benign angiomatous nodules or plaques, often multiple and grouped; usually located in the head and neck region, especially around the ears
- ▪
May be painful, pruritic or pulsatile, and often recur after excision
- ▪
Characterized histologically by proliferations of capillary-sized vessels with epithelioid endothelial cells surrounding larger, thick-walled vessels, accompanied by eosinophils and lymphocytes
- ▪
Associated with arteriovenous shunts and sometimes trauma
- ▪
Kimura disease now thought to be a separate clinicopathological entity
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


