Abstract
Vascular malformations result from errors in the morphogenesis of embryonic capillary, venous, arterial, and/or lymphatic channels. The clinical appearance, natural history, associated manifestations, evaluation, and therapy depend on the type(s) of vessels affected and the extent of involvement of the skin and other tissues. Vascular malformations are lifelong and may worsen over time, potentially creating cosmetic consequences and functional impairment. In addition, they are occasionally found together with other abnormalities as a part of a complex syndrome. Insights into the genetic defects underlying vascular malformations have improved our understanding of the pathways that regulate vascular morphogenesis and provide the basis for development of targeted treatments.
Keywords
vascular malformation, capillary malformation, nevus simplex, telangiectasia, venous malformation, glomuvenous malformation, lymphatic malformation, primary lymphedema, arteriovenous malformation, port-wine stain, cutis marmorata telangiectatica congenita, overgrowth syndrome, Sturge–Weber syndrome, Klippel–Trenaunay syndrome, Proteus syndrome, PTEN hamartoma tumor syndrome, PIK3CA-related overgrowth spectrum, CLOVES syndrome, CLAPO syndrome, megalencephaly–capillary malformation syndrome, Parkes Weber syndrome, Maffucci syndrome, Gorham–Stout disease, hereditary hemorrhagic telangiectasia, blue rubber bleb nevus syndrome, Cobb syndrome, capillary malformation–arteriovenous malformation, Bonnet–Dechaume–Blanc syndrome
▪ Vascular birthmarks; mature angiomas [misnomer]; angiodysplasias ▪ Capillary malformation (CM): port-wine stain, nevus flammeus ▪ Venous malformation (VM): cavernous angioma [misnomer], cavernous hemangioma [misnomer], phlebectasia ▪ Lymphatic malformation (LM): lymphangioma, lymphangioma circumscriptum, lymphangioma simplex, cystic hygroma, cavernous lymphangioma ▪ Arteriovenous malformation (AVM): cirsoid aneurysm, cirsoid hemangioma
- ▪
Although both were once called “angiomas”, vascular malformations differ from the vascular tumors seen in infants and children
- ▪
In slow-flow malformations, capillaries, veins, and/or lymphatic channels are anomalous; most lesions are either apparent at birth or become evident within the first few months or years of life
- ▪
In fast-flow malformations, there is arteriovenous shunting; some lesions are apparent at birth, but the majority become evident later in childhood or adulthood
- ▪
Vascular malformations are lifelong and may worsen over time, creating cosmetic consequences and functional impairment
- ▪
Vascular malformations are occasionally found together with other abnormalities as a part of a complex syndrome
Introduction
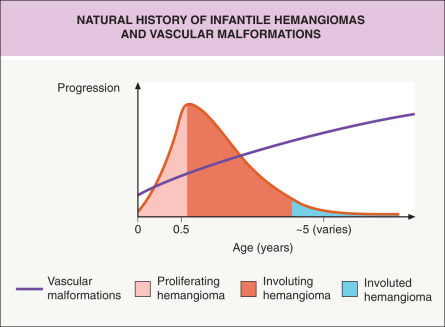
According to the classification system adopted by the International Society for the Study of Vascular Anomalies (ISSVA), there are two types of vascular anomalies: vascular tumors (most commonly the infantile hemangioma) and vascular malformations ( Table 104.1 ; see Table 103.2 ). Vascular malformations are localized defects of vascular morphogenesis, likely caused by dysfunction in pathways regulating the formation of vascular channels during embryonic development. True angiogenesis may occur in some of these lesions, potentially explaining their propensity to thicken and even expand over time. However, in general, vascular malformations have a quiescent endothelium, and they do not exhibit the markers of endothelial cell proliferation seen in infantile hemangiomas during their proliferative phase ( Fig. 104.1 ). Because vascular malformations are not truly proliferating lesions with cellular hyperplasia, the suffix “oma” (meaning “tumor”) has been deemed inaccurate. Thus, the terms “angioma”, “lymphangioma”, and “hemangioma” should not be used for vascular malformations.
| DIFFERENCES BETWEEN VASCULAR MALFORMATIONS AND INFANTILE HEMANGIOMAS | ||
|---|---|---|
| Characteristics | Vascular malformations | Infantile hemangiomas |
| Clinical features | CM: red, macular during infancy VM: blue, compressible, fills with dependency LM: vesicles or large cysts AVM: four stages:
| Premonitory markers at birth in 50%: red telangiectatic, bluish bruise-like, or white/anemic macules Superficial hemangioma: bright red, “strawberry”-like papules and plaques (during proliferative phase) Deep hemangioma: warm, rubbery mass under normal-colored or bluish skin |
| Sex prevalence | Female : male ratio 1 : 1 | Female : male ratio 2 : 1 to 5 : 1 |
| Natural history | Usually evident at birth Lifelong persistence, with commensurate growth during childhood and often slow worsening over time AVM: may worsen with puberty, trauma, and pregnancy | Precursor lesion or no evidence at birth Postnatal proliferation over 5–9 months, then slow involution over years Reach stable involuted stage |
| Radiology | VM or LM (slow flow): hypersignal on MRI T2-weighted sequence VM: thrombi and phleboliths (MRI, CT); gadolinium enhancement on MRI AVM (fast flow): flow voids on MRI T1- and T2-weighted sequences; arteriovenous shunts on Doppler ultrasound | Well-defined mass Some flow voids on MRI T1-weighted sequences (high-flow vessels) |
| Pathology (see Fig. 104.23 ) | CM: ectatic capillaries VM or LM: distorted, interconnected malformed channels AVM: AV fistulae and increased capillaries Typically no increase in cellular turnover | Dense lobular proliferation of endothelial cells forming capillaries with tiny lumens Increased cellular turnover in proliferating lesions can be demonstrated by elevated levels of markers of proliferation (PCNA, Ki-67), ECM remodeling (collagenase, urokinase), and vascular growth (bFGF, VEGF) |
| Immunophenotype | GLUT1-negative | GLUT1-positive |
| Hematology | VM or LVM: chronic LIC, occasional DIC Treatment of flares with low-molecular-weight heparin Possible benefit of low-dose aspirin as prophylaxis | (Kasabach–Merritt phenomenon is not linked to infantile hemangiomas, but to other vascular tumors such as kaposiform hemangioendotheliomas and tufted angiomas, both of which are GLUT1-negative; see Ch. 114 ) |
| Molecular biology | CMs: somatic GNAQ (including SWS) or less often GNA11 mutation Sporadic VMs: somatic TEK (~50%) or PIK3CA (~25%) mutation LMs: somatic PIK3CA mutation Familial vascular malformations and overgrowth syndromes: see Tables 104.2 and 104.5 | Rare families with autosomal dominant inheritance linked to 5q, and somatic loss of heterozygosity of 5q in some hemangiomas Germline mutation in VEGFR2 or TEM8 in some patients Somatic mutation in VEGFR2 or FLT4/VEGFR3 (5q34) in some hemangiomas Clonality in some hemangiomas |
In the current ISSVA classification system , which was updated in 2014, vascular malformations are categorized depending on the predominant type(s) of anomalous vessels:
- •
capillary (CM), classically a port-wine stain (PWS), but also including telangiectasias, cutis marmorata telangiectatica congenita (CMTC), and nevus simplex
- •
venous (VM): a misnomer for this entity is “cavernous hemangioma”, a term with a long history of use to describe deep infantile hemangiomas
- •
lymphatic (LM): this category includes microcystic and macrocystic lesions, which were previously termed lymphangioma and cystic hygroma, respectively
- •
arteriovenous (AVM): fast-flow lesions characterized by malformed arteries, veins, and capillaries with direct communications that produce arteriovenous shunting.
Vascular malformations may occur in any body part or organ system. They are most easily identified in the skin and mucous membranes, with some of these lesions invading deep into underlying muscles, bones, or joints. Others are found in visceral locations. Vascular malformations can appear as localized lesions of varying sizes, occur within a segmental distribution, or be dispersed as multifocal or disseminated lesions.
Vascular malformations are persistent and some worsen over time if not treated. Categorizing a lesion as a CM, LM, VM, AVM, or combined lesion is important, since diagnostic modalities as well as treatment differ depending on the type(s) of vessels involved . In many patients, a multidisciplinary team is required to provide optimal management. Cure is rarely achievable. A realistic aim is improvement and minimization of cosmetic or functional consequences. Many patients require longitudinal care from infancy through adulthood .
History
In the medical literature addressing vascular malformations, lesions and syndromes were originally delineated based on clinical features. Many authors played a major role in their description, including Virchow, Trélat, Monod, Klippel, Trenaunay, Weber, Sturge, Bockenheimer, Maffucci, Bell, and Malan. Unfortunately, pathologists and clinicians did not work together. Consequently, multiple nomenclatures became an obstacle to communication among physicians treating these patients. In 1976, John B. Mulliken (a plastic surgeon in the US), Anthony E. Young (a vascular surgeon in the UK), and Jean-Jaques Merland (a neurointerventional radiologist in France) joined forces to convene the first of many Workshops on Vascular Anomalies. The ISSVA, created in 1992, is dedicated to analyzing the clinical, radiologic, biologic, and pathologic characteristics of vascular lesions and improving diagnosis and treatment via multidisciplinary and international collaboration.
Epidemiology
Little is known regarding the exact prevalence of vascular malformations in the general population. The most common types are CMs, then VMs. The least common are AVMs. Vascular malformations have no gender predilection. They appear to be less frequent in individuals of African and Asian descent.
Pathogenesis
An initial embryologic process, vasculogenesis, creates the primitive vascular plexus. Then angiogenesis, the secondary sprouting of mesoderm-derived endothelial cells, forms new vessels from existing ones, thereby generating most blood and lymphatic vessels. Endothelial differentiation, recruitment of smooth muscle cell precursors to ensheathe endothelial cells and build vessel walls, and, finally, changes in channel size, morphology, and rheology create capillaries, veins, and arteries.
Vascular malformations constitute a heterogeneous group of disorders that result from alterations in formation of blood or lymphatic channels. Dysfunction in signaling processes that regulate migration, differentiation, maturation, adhesion, and survival of the cells of vascular walls is thought to have a pathogenic role. Markers of cellular proliferation are not elevated in vascular malformations. In the cephalic region of the embryo, mural cells associated with endothelial cells come from the neural crest; therefore, a vascular malformation complex involving the head, such as Sturge–Weber syndrome (SWS), is likely caused by a somatic mutation in the embryo’s anterior neural crest or adjacent cephalic mesenchyma. Identification of the genetic defects underlying various types of vascular malformations and the functions of the encoded proteins has provided insights into regulatory pathways critical to vascular morphogenesis ( Table 104.2 ) . Sporadic vascular malformations may be caused by somatic mutations in the same or different genes than those implicated in the less common familial forms of the same type of malformation. For example, somatic TEK mutations that cause constitutive activation of the endothelial cell tyrosine kinase receptor TIE-2 are found in lesional tissue (but not the peripheral blood) in half of patients with sporadic VMs, while germline gain-of-function TEK mutations cause familial cutaneous and mucosal VMs (VMCM) . Advances in our understanding of the pathogenesis of vascular malformations may provide the basis for targeted treatment strategies .
| VASCULAR ANOMALIES FOR WHICH THE MOLECULAR BASIS IS KNOWN | ||||
|---|---|---|---|---|
| Vascular malformation | Mode of inheritance | Mutated gene(s) | Type of mutation | Protein and function |
| Capillary and/or arteriovenous malformations | ||||
| Port-wine stain/Sturge–Weber syndrome | Mosaic, with somatic mutations in lesional tissue | GNAQ | Activating | Q-class G protein α-subunits |
| Port-wine stain/phakomatosis pigmentovascularis type II or overgrowth of an extremity | GNA11 | |||
| Ataxia–telangiectasia | AR | ATM | Loss-of-function | ATM protein is similar to phosphoinositol-3 kinase – regulates cell cycle, DNA repair, p53 |
| Capillary malformation–arteriovenous malformation | AD | RASA1 EPHB4 | Loss-of-function | p120-Ras-GAP protein and EPH receptor B4 in endothelial cells – interact and modulate MAPK signaling by growth factor receptors |
| Cerebral capillary malformations (CCM; familial cerebral cavernomas) * | AD | CCM1: KRIT1 CCM2: CCM2 CCM3: PDCD10 | Loss-of-function | KRIT-1 interacts with KREV-1, RAP-1A and malcavernin (the CCM2 gene product) – roles in MAPK and integrin signaling |
| Hereditary hemorrhagic telangiectasia (HHT; Osler–Weber–Rendu) | AD | HHT1: ENG HHT2: ACVRL1/ALK1 HHT5: GDF2 | Loss-of-function | Endoglin and activin A receptor-like type 1 are two TGF-β receptors with roles in vessel wall integrity, which are activated by GDF2 |
| Juvenile polyposis with HHT | AD | SMAD4 | Loss-of-function | SMAD4 tumor suppressor protein – role in TGF-β signaling |
| Microcephaly-capillary malformation syndrome ** | AR | STAMBP | Loss-of-function | STAM-binding protein, a deubiquitinating enzyme |
| Venous malformations | ||||
| Cutaneous and mucosal venous malformations (VMCM) | AD; mosaic, with somatic mutations in lesional tissue | TEK | Gain-of-function | TIE-2, an endothelial cell-specific tyrosine kinase receptor that binds angiopoietins |
| Blue rubber bleb nevus syndrome | Mosaic, with double (cis) somatic mutations in lesional tissue | |||
| Venous malformations | Mosaic, with somatic mutations in lesional tissue | TEK PIK3CA | Gain-of-function | See above for TIE-2; PIK3CA Activates the AKT/mTOR pathway |
| Glomuvenous malformations | AD with “second hit” in lesional tissue | GLMN | Loss-of-function | Glomulin is a component of a multiprotein complex – role in vascular morphogenesis |
| Lymphatic anomalies | ||||
| Lymphatic malformations | Mosaic, with somatic mutations in lesional tissue | PIK3CA | Gain-of-function | See above for TIE-2; PIK3CA activates the AKT/mTOR pathway |
| Milroy disease (congenital lymphedema) | AD > AR | FLT4 | Loss-of-function | VEGFR-3 – a tyrosine kinase receptor in lymphatic vessels |
| Milroy-like disease | AD | VEGFC | Loss-of-function | VEGF-C – a ligand for VEGFR-3 |
| Lymphedema–distichiasis | AD | FOXC2 | Loss-of-function | Forkhead family transcription factor C2 |
| Hypotrichosis–lymphedema–telangiectasia syndrome | AR, AD | SOX18 | Loss-of-function or dominant-negative | SRY-box 18 transcription factor |
| Hennekam lymphangiectasia–lymphedema syndrome ‡ | AR | CCBE1 FAT4 | Loss-of-function | Collagen & calcium-binding EGF domain-containing protein 1; FAT atypical cadherin 4 |
| Hereditary lymphedema | AD | GJC2 | Loss-of-function | Connexin 47 in gap junctions of lymphatic vessels |
| Oculo-dento-digital dysplasia | AD | GJA1 | Missense | Connexin 43 in gap junctions |
| Lymphedema–choanal atresia | AR | PTPN14 | Loss-of-function | Protein tyrosine phosphatase that may interact with VEGFR-3 |
| Microcephaly, lymphedema, chorioretinal dysplasia syndrome | AD | KIF11 | Loss-of -function | Kinesin family member 11 |
| Arteriopathy | ||||
| CADASIL | AD | NOTCH3 | Loss-of-function in a subset of cases | Accumulation of NOTCH-3 protein in vascular smooth muscle cells |
* Cutaneous vascular malformations, including hyperkeratotic capillary–venous malformations (associated with KRIT1 mutations) and venous malformations (less common), occur in ~10% of patients.
** Features multiple small capillary malformations, hypoplastic distal phalanges, severe intellectual disability, and intractable epilepsy.
‡ Presents with generalized lymphedema, lymphangiectasias (intestinal and pulmonary), facial anomalies (flat face, depressed nasal bridge, hypertelorism), and intellectual disability.
Clinical Features
Most vascular malformations can be correctly categorized based on their clinical features. This helps in selection of the most appropriate investigative tools ( Table 104.3 ), avoiding redundant and unnecessary diagnostic imaging procedures.
| INVESTIGATIVE TOOLS FOR VASCULAR MALFORMATIONS | ||||
|---|---|---|---|---|
| Tool | Vascular lesion | |||
| Capillary malformation | Lymphatic malformation | Venous malformation | Arteriovenous malformation | |
| Ultrasonography | +/− | ++++ | +++ | ++++ |
| CT with contrast | − | ++ | ++ | ++ |
| MRI with gadolinium | − * | +++ (no gadolinium enhancement) | ++++ (gadolinium enhancement) | +++ |
| 3D-angio CT or 3D-angio MRI | − | − | ++ | ++++ |
| Arteriography | − | − | − | ++++ |
| Phlebography | − | − | +/− | − |
| Lymphography (isotopic) | − | +/− | − | − |
| Biopsy | +/− | +/− | − | − |
* For evaluation of possible Sturge–Weber syndrome (SWS), brain MRI with gadolinium or MR susceptibility-weighted imaging (SWI) are the recommended initial imaging modalities.
Capillary Malformations
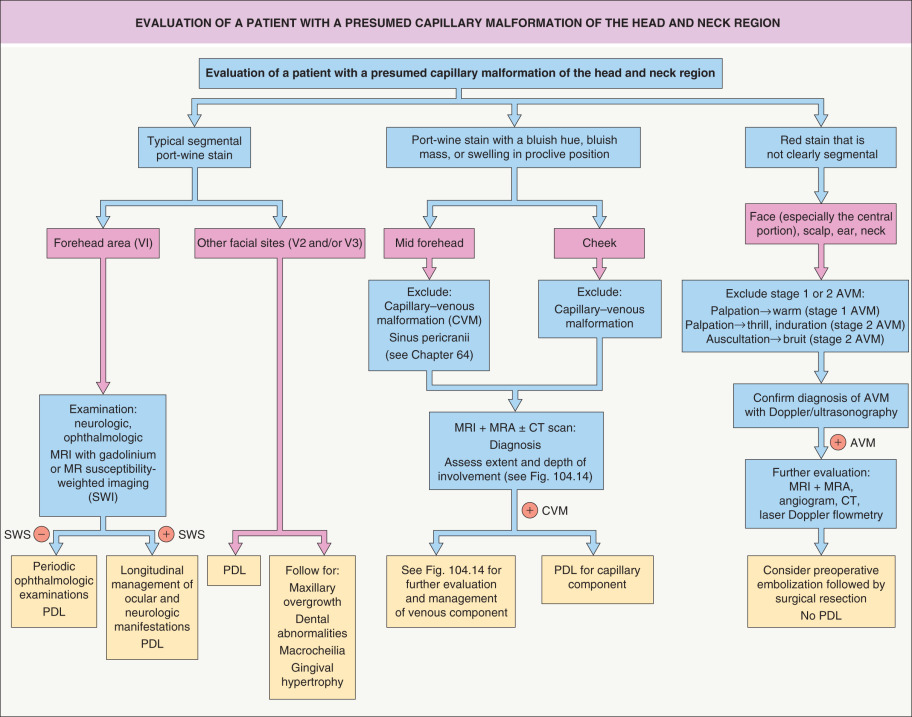
Clinical variants of CM include nevus simplex, PWSs, reticulated CMs, geographic CMs, CMTC, and telangiectasias . Occasionally, these lesions represent the most obvious sign of a complex syndrome. An approach to the evaluation of a patient with a presumed CM of the head and neck region is shown in Fig. 104.2 .
Nevus simplex
Nevus simplex (salmon patch) is a very common congenital vascular stain evident in 30–80% of neonates . Although classified as a CM by the ISSVA, nevus simplex is thought to represent remnants of the fetal circulation rather than a mosaic condition due to a somatic mutation. These pink-to-red macules and patches with somewhat indistinct borders have characteristic locations on the forehead/glabella in a V-shape (“angel kiss”), eyelids, philtrum, occiput, nape (“stork bite”), and lumbosacral area ( Fig. 104.3A&B ). They often become more prominent with crying or vigorous activity. The term nevus simplex complex has been proposed for extensive lesions .
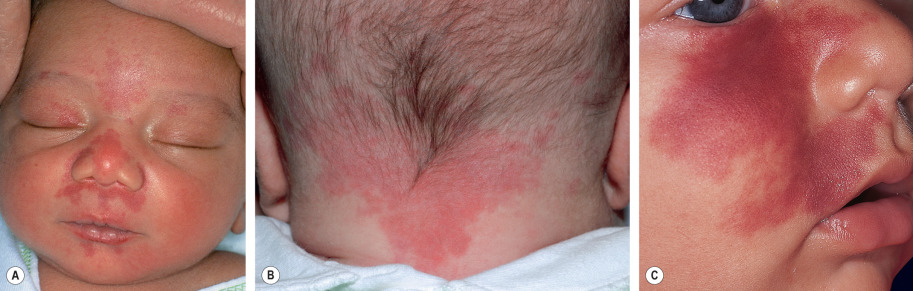
Facial nevus simplex tends to fade spontaneously between 1 and 3 years of age, but extrafacial and some glabellar lesions are more persistent. During infancy, eczematous dermatitis may preferentially or solely develop within a nevus simplex. The vast majority of children with a nevus simplex do not have associated abnormalities; however, a prominent or persistent midfacial nevus simplex is a feature of several syndromes, including Beckwith–Wiedemann and megalencephaly–CM. Although there has been controversy regarding whether a lumbosacral nevus simplex represents a possible sign of occult spinal dysraphism, most authors do not recommend spinal imaging in the absence of additional cutaneous findings (see Ch. 64 ) .
Port-wine stain and variants
A somatic activating mutation in GNAQ (Q-class G protein α-subunit gene) in affected skin (especially the blood vessels) and regional extracutaneous tissues (e.g. brain, eyes) underlies isolated PWSs and Sturge–Weber syndrome (SWS), respectively. This mutation stimulates mitogen-activated protein kinase (MAPK) signaling, which results in increased cell proliferation and decreased apoptosis .
PWSs typically present at birth as well-demarcated, bright or deep red macules and patches, with their color resembling that of port wine. Fainter red or pink stains have been termed “ nevus roseus ” because their color resembles that of rosé wine . Some light red–pink stains are not solid but finely reticulated, often with a blotchy appearance and indistinct borders; these reticulated CMs are distinct from CMTC, which has a more well-defined, purplish net- or tram track-like pattern.
The growth of PWSs is commensurate with the child’s growth. They can be localized, have a segmental pattern, or be multifocal and widespread. The distribution patterns of facial PWSs are thought to reflect the prominences that form during embryonic craniofacial development and their associated vasculature; because these regions resemble the dermatomes of the trigeminal nerve, the following areas are classically recognized: V1 – forehead and upper eyelid; V2 – maxillary region ( Fig. 104.3C ); and V3 – mandibular region.
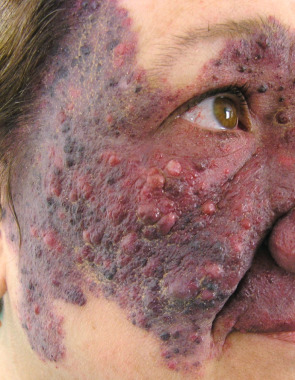
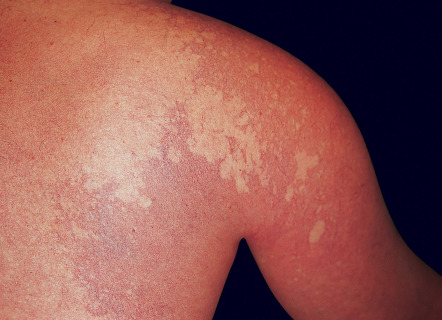
Over time, PWSs, especially those in the maxillary and mandibular areas, often develop a deeper red hue, changing from pinkish-red at birth to purplish-red by adulthood. Affected skin may thicken and become nodular ( Fig. 104.4 ), and superimposed pyogenic granulomas occasionally appear. In a study of 173 patients with PWSs, thickening was observed in 11% (median age 32 years), nodularity in 24% (median age 44 years), and both in 6% (median age 45 years) . These changes are rarely observed in the lighter “nevus roseus” or in PWSs located on the trunk and limbs. Overgrowth of the soft tissues and facial bones underlying a PWS can also occur, creating problems such as an open-bite deformity. Affected gums and lips may enlarge, potentially resulting in epulides, gingival bleeding, macrocheilia, and lip incompetence. As with nevus simplex, eczematous dermatitis may have a predilection for areas of skin affected by the PWS.
PWSs are congenital in the vast majority of patients. However, acquired PWSs have been described in adolescents and adults, and onset of such lesions may be precipitated by trauma. Of note, early morphea, especially the linear variant, occasionally presents with a red vascular patch that mimics an acquired PWS .
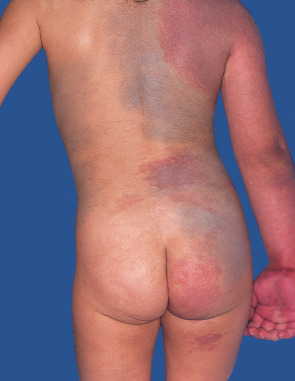
A CM can be admixed with a network of blanched macules representing a nevus anemicus ( Fig. 104.5 ). The association of a CM with aberrant Mongolian spots (dermal melanocytosis) ( Fig. 104.6 ) or a nevus spilus (speckled lentiginous nevus) is referred to as phakomatosis pigmentovascularis (PPV) ( Table 104.4 ) . Patients with PPV type II have the same activating mutation, either in GNAQ or GNA11 , in both their port-wine stain and dermal melanocytosis.
| PHAKOMATOSIS PIGMENTOVASCULARIS (PPV) | ||||
|---|---|---|---|---|
| Type | Descriptive name | Proportion of reported cases | Major cutaneous components | Other reported cutaneous components |
| Ia/b | * | <5% * | Epidermal nevus + PWS | |
| IIa/b | Phakomatosis cesioflammea | 75% | Dermal melanocytosis (aberrant Mongolian spots) + PWS | Nevus anemicus, hypotrichosis, lipohypoplasia, hypoplastic nails |
| IIIa/b | Phakomatosis spilorosea or melanorosea | 10% | Nevus spilus (macular type) + light red–pink CM (“nevus roseus”) | Nevus anemicus, lymphedema, hypotrichosis |
| IVa/b | Phakomatosis melanovascularis or unclassifiable | 10% | Café-au-lait macule + CM Variable, sometimes with features of II + III or II + V | Nevus anemicus, nevoid hyper- or hypopigmentation, nevus sebaceus |
| Va/b | Phakomatosis cesiomarmorata | <5% | Dermal melanocytosis + cutis marmorata telangiectatica congenita | |
* Existence as a type of PPV has been challenged considering that epidermal nevi are not melanocytic lesions; epidermal nevi can occur together with vascular malformations in Proteus, PTEN hamartoma tumor, and CLOVES syndromes (see Table 104.5 ), which are not generally considered as PPV.
Sturge–Weber syndrome
SWS is a sporadic neurologic disorder in which a facial PWS is associated with ipsilateral leptomeningeal/brain and ocular vascular anomalies. Although all three components constitute a diagnosis of “complete” SWS, patients with a PWS plus CNS involvement alone are also typically diagnosed as having SWS, whereas those with a facial PWS plus ocular involvement alone are often classified separately. The brain and/or ocular manifestations of SWS rarely occur without an associated PWS. Mosaicism due to a somatic activating mutation in GNAQ represents the cause of both SWS and non-syndromic facial PWSs (see above), with the former resulting from a mutation that arises earlier in development and affects the vasculature of the eye and CNS as well as the skin .
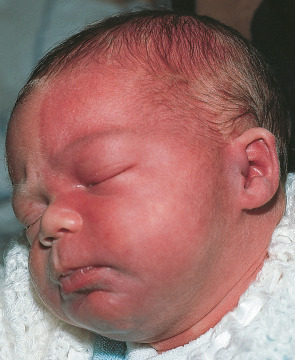
Facial PWSs associated with SWS have classically been considered to involve the V 1 region, which includes the forehead and upper eyelid , often with extension unilaterally or bilaterally over more of the face ( Fig. 104.7 ). Recent studies have shown that involvement of the “forehead area”, which includes the upper eyelid and is delineated inferiorly by a line drawn from the outer canthus to the top of the ear, represents the best clinical predictor for SWS ; this area encompasses portions of V 2 /V 3 as well as V 1 and is thought to correspond with the embryonic frontonasal prominence . The latter includes neural crest-derived vasculature and develops in conjunction with the forebrain, which gives rise to the cerebral cortex and optic vesicles. A somatic GNAQ mutation occurring in the frontonasal area prior to migration of embryonic neural crest derivatives, which also include the leptomeninges and choroid, may therefore explain the forehead location of CMs associated with cerebral and ocular vascular malformations. Some SWS patients also have PWSs on the extremities and trunk. In addition, SWS occasionally occurs in the setting of PPV type II (see Table 104.4 ).
Ocular involvement in SWS can result in enlarged venous vessels affecting the conjunctiva, episclera, retina, and/or choroid. The most common ocular manifestation is glaucoma, which affects 30–60% of patients with a PWS on the forehead and/or eyelids; choroidal hemorrhage and retinal detachment are rare complications. Ocular involvement is more frequent when the PWS affects both the V 1 and V 2 regions. Glaucoma may be detected at birth because of buphthalmos (enlargement of the globe), and acute glaucoma with a cloudy cornea can be an infantile emergency. Usually, however, increased eye pressure develops slowly. As a result, glaucoma may become evident during late childhood, adolescence, or even adulthood. Thus, periodic lifelong assessment of visual function and pressure of both eyes, as contralateral glaucoma occasionally occurs, is mandatory.
Neurologic symptoms can result from hypoperfusion of the ipsilateral brain due to CVMs within the pia mater, absence of superficial cortical veins, and dilated deep draining veins. Over time, chronic hypoxia may lead to cerebral hemiatrophy and calcifications, with the occipital region being the most frequent location. The most common neurologic manifestation is seizures, which are most often focal motor, either affecting the side of the body opposite to the vascular anomaly or generalized. The seizures typically develop in the first two years of life and affect ≥75% of children with SWS ; they may be difficult to control with anticonvulsants, especially early on. Additional findings can include stroke-like episodes with contralateral hemiparesis or hemiplegia; developmental delay affecting motor and cognitive skills; emotional and behavioral problems; attention deficit disorder; and migraine headaches. Endocrine dysfunction such as central hypothyroidism and growth hormone deficiency may also occur, even in patients with normal neuroimaging .
The risk of ocular and/or neurologic manifestations of SWS is ~25–50% when a unilateral PWS affects the full V 1 distribution or most of the forehead area; it is lower when the PWS involves a smaller portion of this area and higher (>50%) for a bilateral PWS covering the forehead and both upper eyelids . In a study of 55 patients with the leptomeningeal vascular anomalies of SWS, the associated cutaneous CM was unilateral in 63.5%, bilateral in 31%, and absent in 5.5% .
Neuroimaging should be considered in any child with a facial PWS involving the forehead area (see above) in order to identify and delineate the extent of CNS abnormalities (see Fig. 104.2 ). MRI with gadolinium contrast or magnetic resonance susceptibility-weighted imaging (SWI), which does not require contrast administration, are currently the recommended modalities and can demonstrate early leptomeningeal changes with enlargement of transmedullary and periventricular veins. In asymptomatic patients at risk of SWS, such imaging is insensitive in the first few months of life but by ≥1 year of age can reliably exclude leptomeningeal involvement. Contrast-enhanced fluid attenuation inversion recovery (FLAIR) imaging and high-resolution blood oxygen level dependent (BOLD) magnetic resonance venography (MRV) may improve detection of leptomeningeal disease when compared with routine contrast-enhanced T1-weighted MRI. Gyriform calcifications and atrophy develop during childhood in SWS patients and are visible by CT scans and other neuroimaging modalities.
Early diagnosis of SWS is important, as prophylactic aspirin administration may reduce the frequency of stroke-like episodes and seizures . Some infants and children with SWS have cognitive deficits that are more severe than anticipated based upon their limited cortical involvement on conventional MRI studies. Functional cerebral imaging, e.g. SPECT (single photon emission computed tomography) that evaluates regional cerebral blood flow or PET (positron emission tomography) that demonstrates metabolism of glucose, can provide additional prognostic information. Quantitative electroencephalography (EEG) may also aid in screening for brain involvement in asymptomatic infants at risk of SWS .
Capillary malformations associated with overgrowth
PWSs and reticulated CMs on the trunk and extremities may be associated with regional overgrowth and other syndromic features ( Table 104.5 ) . Mosaicism for mutations that result in activation of the phosphatidylinositol 3-kinase (PI3K)/AKT pathway have been found to underlie several of these conditions (see Figs 55.3 and 113.2 ). The various terms and eponyms used for overgrowth syndromes are likely to be refined in the future as the molecular etiologies are further clarified.
| OVERGROWTH SYNDROMES WITH VASCULAR MALFORMATIONS | ||||||
|---|---|---|---|---|---|---|
| DCMO | PIK3CA -Related Overgrowth Spectrum | Proteus | PHTS | |||
| M-CM | KTS | CLOVES | ||||
| Gene | ? GNA11 (in some patients) | PIK3CA, mosaic | PIK3CA * , mosaic | PIK3CA , mosaic | AKT1 , mosaic | PTEN germline with somatic second mutation |
| Asymmetric/segmental overgrowth | ||||||
| At birth | + | + | + | + | −/minimal | + |
| Progressive | − | − | + | + | +++ | + |
| Correlates with site(s) of vascular malformation | − | − | + | + | − | variable |
| Digital abnormalities | 30%: syndactyly, sandal-gap, macrodactyly | Syndactyly > polydactyly, macrodactyly; sandal gap | Occasional macrodactyly, syndactyly (associated with deep venous anomalies) | Broad hands/feet, macrodactyly (especially 3 rd toe), sandal gap | Macrodactyly with distorted/deformed digits | − |
| Macrocephaly | − | +; (hemi)megalencephaly, dolichocephaly | − | 30% with (hemi)megalencephaly | Dolichocephaly | + |
| Capillary malformations | Reticulated ± confluent areas; widespread, block-like | Reticulated; widespread, block-like; persistent nevus simplex | Geographic, well-demarcated, dark (associated with LM) or blotchy light; on extremity ± extension | Geographic, well-demarcated, dark; on trunk overlying lipomatous mass | Well demarcated; often on trunk | Variable |
| Venous anomalies | Variable prominent veins | Variable prominent veins | VM § ; often persistent lateral marginal vein, abnormal deep venous system | VM § of superficial, deep thoracic, and often major central veins | Variable VM § | Variable VM; dilated draining veins associated with high-flow lesions |
| Lymphatic anomalies | − | − | Often LM with overlying vesicles, lymphedema | Truncal LM within lipomatous mass; overlying vesicles | Variable LM | Variable LM, lymphedema |
| Arteriovenous malformations | − | − | − | Variable spinal/paraspinal AVM | − | Fast-flow “PTEN hamartoma of soft tissue” with intramuscular involvement and ectopic fat |
| Other cutaneous features | − | Hyperelastic, soft/doughy skin | − | Lipomatous truncal masses, epidermal nevi, furrowed soles | Cerebriform connective tissue nevi of palms/soles, epidermal nevi, lipomatous overgrowth, regional lipohypoplasia | Lipomatosis, epidermal nevi, pigmented macules of genitalia; later tricholemmomas, acral keratoses, oral papillomas, neuromas, sclerotic fibromas |
| Other extracutaneous features | − | Polymicrogyria, ventriculomegaly, cerebellar tonsillar ectopia, hypotonia, seizures, developmental delay; frontal bossing, joint hypermobility | − | Variable polymicrogyria, seizures, and other features of M-CM | Lung bullae; hyperostoses, megaspondylodysplasia with scoliosis; long face with ptosis, depressed nasal bridge, anteverted nares, and open mouth at rest | See Table 63.3 |
| Tumors | − | Rare Wilms tumor ** | − | Rare Wilms tumor ** | Parotid monomorphic adenoma, ovarian cystadenoma, testicular tumors, meningioma | Increased risk of thyroid, breast, endometrial, renal, and GI carcinomas |
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


