The Neurofibromatoses: Introduction
|
|
Epidemiology
The “modern age” of neurofibromatosis began in 1981, with Riccardi’s detailed clinical descriptions of the features and natural history of von Recklinghausen disease and the subsequent description of “central neurofibromatosis with bilateral acoustic neuroma.” With the availability of gene-based diagnosis, several distinct clinical syndromes have been identified: (1) neurofibromatosis type 1 (NF-1), also known as von Recklinghausen disease; (2) neurofibromatosis type 2 (NF-2), whose cardinal feature is the development of bilateral vestibular schwannomas; (3) segmental or mosaic NF-1; (4) Legius syndrome (SPRED1 mutation) leading to autosomal dominant transmission of café-au-lait spots, intertriginous freckling and macrocephaly; and (5) schwannomatosis. Both NF-1 and NF-2 occur equally in all ethnic groups. NF-1 occurs at an incidence of 1 in 3,000 live births, whereas NF-2 is much less common with an estimated incidence of 1 in 40,000 live births.
Etiology and Pathogenesis
NF-1 is inherited in an autosomal dominant fashion. Although the expressivity of NF-1 varies considerably, even among individuals in the same family who are genotypically identical, the disorder is considered to be 100% penetrant. Thus, apparent skipping of NF-1 between generations may be the result of misdiagnosis, nonpaternity, or the occurrence of a new mutation in the grandchild. Although a large number of NF1 mutations have been described, only two genotype-phenotype correlations have been noted. Individuals who have deletions of the entire NF1 gene have an increased risk of facial dysmorphism, mental retardation, early appearance of neurofibromas, and the presence of plexiform neurofibromas.1 In addition, individuals who have a specific 3 base pair in-frame deletion in exon 17 of the NF1 gene may have café-au-lait spots, intertriginous freckling and Lisch nodules, but do not develop cutaneous, subcutaneous or plexiform neurofibromas. neurofibromas. There is evidence that NF-1 is more severe when inherited from the mother rather than from the father.
The gene NF1 is located on the long arm of chromosome 17, and encodes a protein called neurofibromin. Homozygosity for mutant NF1 alleles has not been reported, probably because it is a lethal condition. NF1 has an unusually high mutation rate, estimated at 2.4 × 10−5 to 10 × 10−5 gametes per generation, one of the highest of known inherited disorders.2 This may reflect the large size of the gene, which spans 350 kb of genomic DNA, and contains 59 exons which encode a peptide containing over 2,800 amino acids. An alternative hypothesis is that there is some structural property of the gene that renders it particularly susceptible to mutation. New mutations account for approximately 50% of cases, and are usually on the paternally inherited allele. In contrast to many other dominant disorders, the frequency of new mutations does not appear to increase with advancing paternal age.
Most NF1 mutations result in reduced intracellular levels of the NF1 gene product, neurofibromin; this appears to be sufficient to cause many of the clinical manifestations of the disease. Tumorigenesis, including the development of benign dermal neurofibromas, appears to be dependent on inactivation of the normal NF1 allele in somatic cells, a process referred to as loss of heterozygosity.3 Loss of heterozygosity is a critical step in tumorigenesis in many inherited cancer predisposition syndromes (e.g., retinoblastoma, Li-Fraumeni syndrome), and the genes involved are called “tumor suppressor genes.” Tumorigenesis is initiated when both copies of the gene cease functioning normally—the first as a result of an inherited mutation, and the second as the result of a second somatic “hit” interfering with the function of the previously normal allele.
Neurofibromin is found in a variety of cell types, including neurons, oligodendrocytes, and nonmyelinating Schwann cells. Considerable evidence has been developed for the role of neurofibromin as a negative regulator of RAS.4,5 The RAS gene family encodes membrane-associated, guanine nucleotide-binding proteins that are involved in the regulation of cellular proliferation, differentiation, and learning. RAS exists in an active (RAS-GTP) and inactive (RAS-GDP) state. By favoring conversion of RAS from its active state to its inactive state, neurofibromin downregulates the downstream effects of RAS, which include promoting learning, memory, synaptic plasticity, and cell growth and proliferation.4,6 Mediators of the downstream effects of RAS include mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K), protein kinase B (PKB), and mammalian target of rapamycin (mTOR) kinase. Spred1 also inhibits activation of the MAPK pathway.
Four different forms of neurofibromin exist, resulting from alternate splicing. The type II variant includes a 21 amino acid sequence encoded by exon 23a, while type I does not. This sequence appears to be critical for RAS regulation and learning. In the brain, there are three major isoforms of RAS; K-RAS has been shown to be the primary target of neurofibromin activity.5 Activation of RAS by neurofibromin stimulates G-protein activity, resulting in activation of adenylyl cyclase. Cyclic AMP and other downstream intermediates appear to play a role in cell growth, learning, and memory. Neurofibromin also associates with microtubules and plays a role in regulating GABA-ergic inhibitory neuronal activity in the hippocampus.7
RAS is a membrane-bound protein, and requires farnesylation (addition of a C15 isoprenoid to a cysteine residue near the C-terminus) for activity, a process that is catalyzed by farnesyltransferase. Agents that inhibit farnesyltransferase cause general inhibition of the RAS pathway, much as neurofibromin does. Farnesyltransferase inhibitors reverse the proliferative phenotype in Nf1-deficient mouse cells, and can reverse spatial learning impairments in a mouse model of NF1, by decreasing RAS levels.6 These observations have prompted investigations into the use of farnesyltransferase inhibitors for the treatment of NF-1-related complications, such as rapidly growing plexiform neurofibromas.
Loss of heterozygosity of the NF1 gene in Schwann cells has been shown to be the necessary event for the development of discrete neurofibromas.8 Furthermore, neurofibromin-deficient Schwann cells secrete a substance which stimulates mast cell migration which, in turn, stimulates production of extracellular matrix and angiogenesis.9 Astrocytes lacking NF1 expression cannot form optic nerve gliomas on their own; a brain environment heterozygous for NF1 is necessary for the development of these tumors.10
Clinical Findings
A consensus development conference was held by the National Institutes of Health in 1987 in order to establish diagnostic criteria to promote better clinical research and care of patients with NF-1.11 The seven diagnostic features recognized at this conference (Table 141-1), and the recommendation that two or more of these seven features be present before a diagnosis of NF-1 is established, have proven extremely useful and continue to be used without modification 20 years later. Perhaps the one caveat is the identification of a separate autosomal dominant syndrome caused by an inactivating mutation of the gene encoding sprouty-related EVH1 domain-containing protein 1 (SPRED1) which leads to the development of café-au-lait spots, intertriginous freckling, and macrocephaly, but none of the other manifestations of NF-1 (Legius syndrome).12
Café-au-lait spots, which are flat, pigmented macules, are often the first manifestation of NF-1 to appear (Fig. 141-1). Frequently present at birth, they become more numerous as the infant grows; new ones may continue to appear throughout the first decade of life. Once noticed, they tend to grow in size in proportion to the overall growth of the child. Although infrequently found on the face, they may be noted anywhere on the body. The size, shape and contour of café-au-lait spots are of no diagnostic significance, and the oft-quoted adage about smooth-edged café-au-lait spots being more typical of NF-1 rather than McCune–Albright syndrome is incorrect (see Section “Differential Diagnosis” and Box 141-1). When café-au-lait spots overlap each other, the area of overlap may be darker than either individual spot. When found within Mongolian spots, they are typically surrounded by a more lightly pigmented halo. Individuals with large numbers of café-au-lait spots do not have “more severe” NF-1, and the location of the macules in no way predicts the location of subsequent neurofibromas.13
Most Likely
|
Less Likely
|
Café-au-lait spots represent collections of heavily pigmented melanocytes of neural crest origin in the epidermis. Although the cells may contain increased numbers of “giant” melanosomes, or melanin macroglobules, these are not unique to NF-1, and their presence or absence in biopsies is not helpful diagnostically.
The diagnostic criterion for NF-1 is six or more café-au-lait spots greater than 5 mm in diameter in prepubertal individuals or 1.5 cm in adults. Of all children ultimately diagnosed with NF-1, 53% will have six or more café-au-lait spots by age 3 years and 97% by age 6 years.14
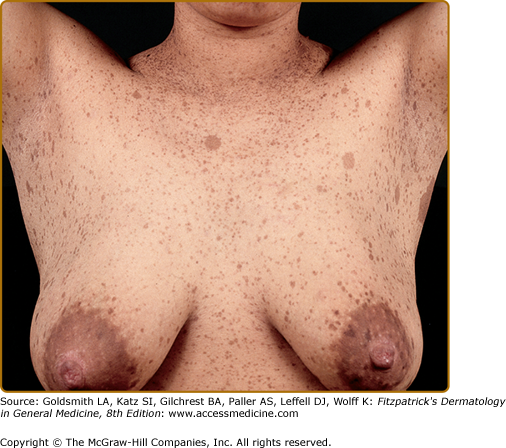
Café-au-lait spots smaller than 5 mm are referred to as freckles, and are commonly present in the axillae, inguinal region, and under the breasts (Fig. 141-2). Unlike ordinary freckles in these locations, these lesions are not related to sun exposure, and are considered virtually pathognomonic of NF-1 (Crowe’s sign). Of all children ultimately diagnosed with NF-1, 81% will have intertriginous freckling by age 6 years.14
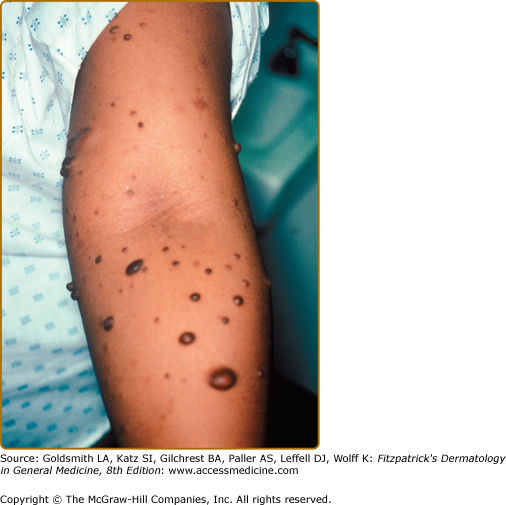
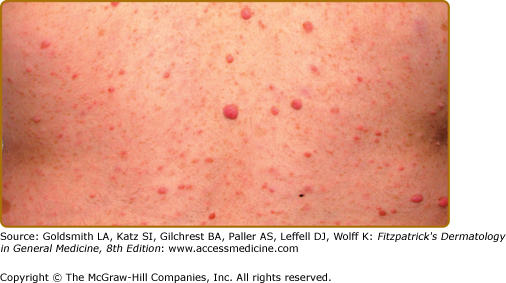
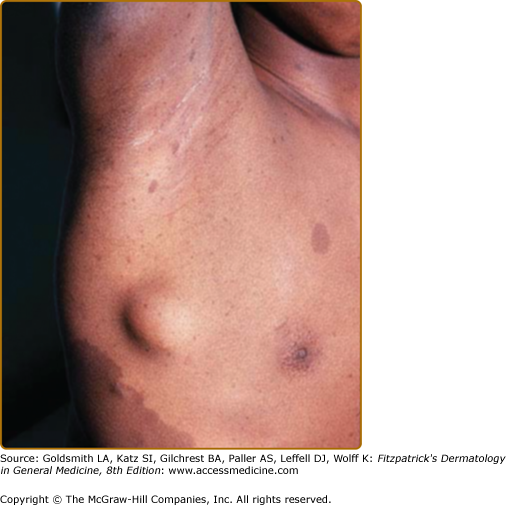
Neurofibromas, which consist of Schwann cells, mast cells, fibroblasts, and perineural cells, are benign nerve sheath tumors that appear as discrete masses arising from peripheral nerves.15 Cutaneous neurofibromas protrude just above the skin surface or lie just under the skin with an overlying violaceous hue (Fig. 141-3). They are softer than the surrounding connective tissue, often creating a “buttonholing” sensation when a finger is rubbed gently over the surface (Fig. 141-4). Subcutaneous neurofibromas that arise from peripheral nerves, both under the skin and deep in the viscera, are generally much harder (Fig. 141-5). If they arise from the dorsal root ganglia, they may grow through neural foramina, compressing the spinal cord, creating a “dumbbell” appearance. Subcutaneous neurofibromas in the neck may feel like a “beaded necklace,” often being confused with lymph nodes. While less than 20% of children under 10 years of age have cutaneous neurofibromas,14 they generally start appearing after puberty and increase in number as the patient grows older. Women who have NF-1 often comment on the appearance of cutaneous neurofibromas during pregnancy. The majority of adult patients with NF-1 probably have numerous asymptomatic deep neurofibromas involving the dorsal roots and other larger nerves. On occasion, neurofibroma-associated pruritus may be severe enough to require treatment with antihistamines.
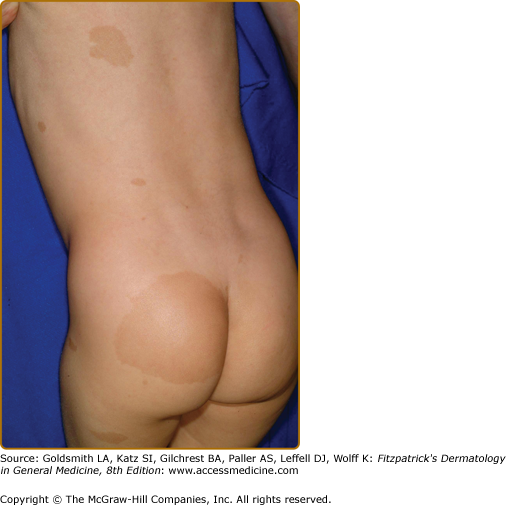
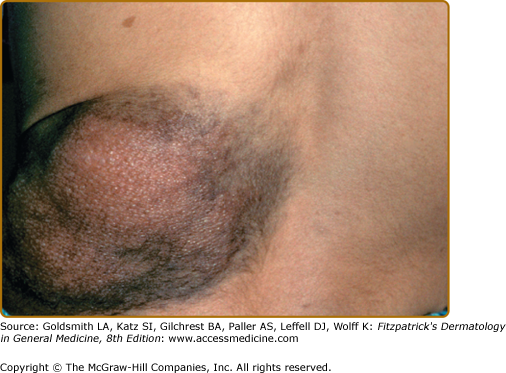
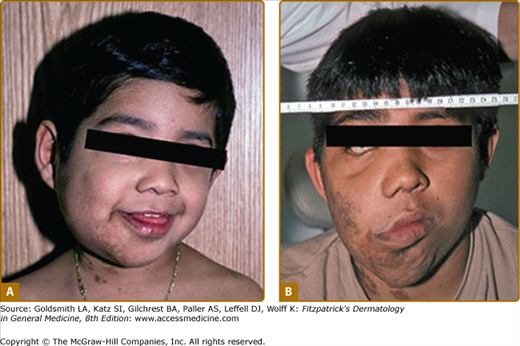
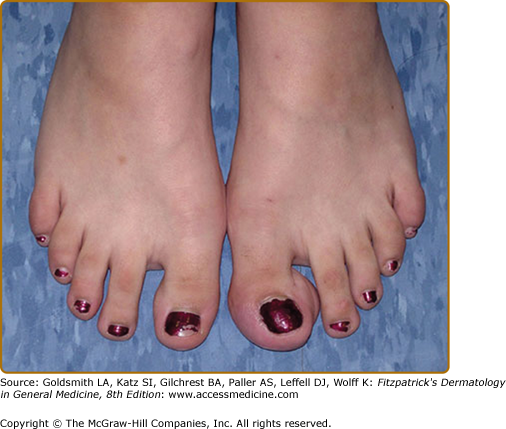
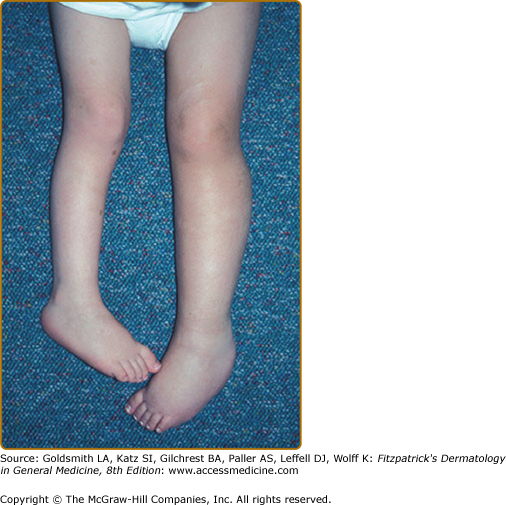
Plexiform neurofibromas, histologically similar to discrete neurofibromas, are benign peripheral nerve sheath tumors which involve single or multiple nerve fascicles, often arising from branches of major nerves.16 They may elicit a “wormy” sensation on palpation, as one feels multiple thickened nerve fascicles. Often there is overlying hyperpigmentation (“giant café-au-lait spot”) or hypertrichosis (Fig. 141-6). Most plexiform neurofibromas are present at birth or become apparent during the first several years of life. Externally visible plexiform neurofibromas are easily identified and may lead to disfigurement, blindness (secondary to amblyopia, glaucoma, or proptosis), or loss of limb function (eFig. 141-6.1, Fig. 141-7, Fig. 141-8). In contrast, thoracic or abdominal plexiform neurofibromas may have no external manifestations but may have equally devastating consequences due to invasion or compression of vital structures (e.g., ureters, bowel, spinal cord, etc.).
The growth rate of plexiform neurofibromas is highly variable. Periods of rapid growth alternating with long periods of quiescence are common. Malignant peripheral nerve sheath tumors, which generally arise from plexiform neurofibromas, may develop silently in deep plexiform neurofibromas and not give rise to symptoms until distant metastases have occurred. While loss of heterozygosity at the NF-1 locus may lead to the formation of benign neurofibromas, the generation of malignant transformation of a benign plexiform neurofibroma may be due to cell cycle regulators beyond the ras oncogene. For example, mice which have null mutations in both the Nf-1 and p53 genes uniformly develop malignant tumors.17,18 Physicians caring for individuals with NF-1 should be alert to development of a malignancy; plexiform neurofibromas should be biopsied if they exhibit rapid growth or cause significant pain or focal neurologic dysfunction.
Approximately 15% of children with NF-1 will develop optic pathway tumors (OPT); however, only half of these patients will ever develop symptoms, giving an overall incidence of symptomatic OPT of 7%.19,20 Recent series of NF-1-associated OPT have identified an approximately 2:1 female predominance of patients, as well as a lower incidence in African-American children. These observations suggest the possibility that either hormonal factors or modifying genes may influence the development of OPT.
The period of greatest risk for the development of symptomatic OPT in NF-1 is during the first 6 years of life. In addition, the development of a symptomatic tumor after age 6 years is extremely unusual.19,20 Thus, physicians caring for children with NF-1 should be sensitive to the signs and symptoms of OPT in this young age group. Approximately 30% of children with symptomatic OPT will present with the rapid onset of proptosis, with moderate to severe visual loss in the affected eye (eFig. 141-8.1). Another 30% of children will have abnormal ophthalmologic examinations, without any visual symptoms, leading to discovery of the tumors. As young children rarely complain of visual loss, even when severe, thorough annual eye examinations are imperative in all young children with NF-1. When present, ophthalmologic signs may include an afferent papillary defect, optic nerve atrophy, papilledema, strabismus, or defects in color vision. Finally, as many as 40% of children who have chiasmal tumors develop precocious puberty. Accelerated linear growth will be the first sign of precocious puberty, underscoring the need for all children with NF-1 to have annual assessments of growth using standard growth charts. Children with chiasmal tumors often have no ophthalmologic symptoms or signs. Early detection of precocious puberty is important as both the accelerated linear growth and the development of secondary sexual characteristics can be can be aborted with the use of a long-acting luteinizing hormone releasing hormone agonist.21
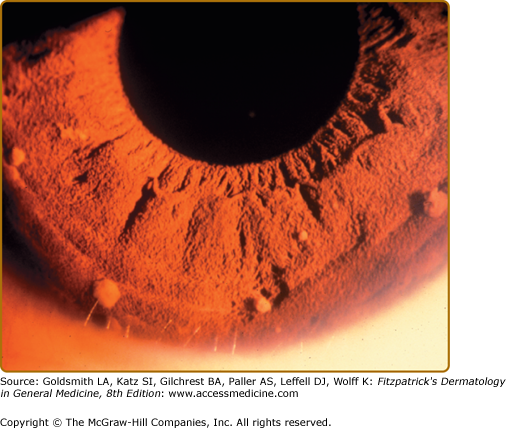
Lisch nodules are slightly raised, well-circumscribed melanocytic hamartomas of the iris thought to be virtually pathognomonic of NF-1 (Fig. 141-9). They are best seen using a slit lamp, which is necessary to distinguish them from the more commonly seen flat iris nevi, which are not associated with NF-1. They do not cause any functional impairment of vision. The frequency with which they are found increases with age; although Lisch nodules are found in over 90% of adults with NF-1, only 30% of children with NF-1 under 6 years of age have them.14
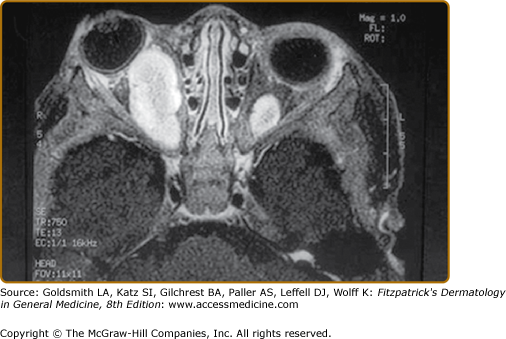
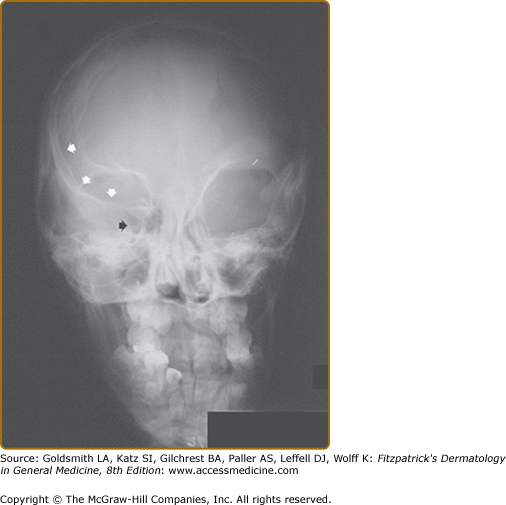
There are two bony lesions distinctive enough to be included in the diagnostic criteria for NF-1. The first, dysplasia of the wing of the sphenoid bone, results in poor formation of the wall and/or floor of the orbit (eFig. 141-9.1). This congenital mesodermal dysplasia may be, but is not always, clinically apparent leading to proptosis (from herniation of meninges or brain into the orbit) or enophthalmos.
eFigure 141-9.1
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


