The hypersensitivity syndromes are a group of disorders mediated by immunologic or hypersensitivity reactions to foreign proteins such as food or drugs, or to infectious agents, immunizations, and malignancies. Although diagnosis of these disorders is often relatively easy, difficulties commonly arise in determining the underlying cause. The most common hypersensitivity reactions are drug reactions, and the skin is a common target. The overall incidence of drug reactions in hospitalized children is 9.5%, and in outpatient children it is 2.5%, with reactivity to antibiotics occurring most often. Urticarial and exanthematous reactions are seen most often. If fever, lymphadenopathy, or facial edema accompanies an exanthematous drug reaction, the possible diagnosis of a systemic hypersensitivity disorder (drug reaction with eosinophilia and systemic symptoms [DRESS]) must be considered. The various types of drug reactions are summarized in Table 20-1 . These include Stevens–Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), lichenoid drug eruptions ( Fig. 20-1 ) (see Chapter 4 ), fixed drug eruptions, pustular eruptions (acute generalized exanthematous pustulosis [AGEP]), acneiform eruptions (see Chapter 8 ), pseudoporphyria (see Chapter 19 ), drug-induced vasculitis (see Chapter 21 ), and drug-induced lupus ( Chapter 22 ). Some children experience multiple antibiotic sensitivity, most commonly to penicillins, sulfonamides, cephalosporins, and macrolides. In these children, the manifestations of drug sensitivity can be different; for example a child may show an exanthematous response to amoxicillin, DRESS after phenytoin, and erythroderma after ingestion of sulfamethoxazole within a period of a few months. The general approach to a child with suspected drug reaction is outlined in Box 20-1 .
| Eruption | Key Drugs | Lesional Pattern | Mucosal Changes |
|---|---|---|---|
| Urticaria | Penicillins, cephalosporins, sulfonamides, tetracyclines, aspirin/NSAIDs, radiocontrast media | Pruritic erythematous wheals | None |
| Angioedema | Aspirin/NSAIDs, ACE inhibitors | Swelling of subcutaneous and deep dermal tissues | May be present |
| Serum sickness-like reaction | Cephalosporins, penicillins, minocycline, sulfonamides, macrolides, rifampin, ciprofloxacin, griseofulvin, itraconazole, bupropion, fluoxetine, rituximab, H1N1 vaccine | Urticarial or erythema multiforme-like | None |
| Exanthematous | Penicillins, sulfonamides, cephalosporins, antiepileptics | Erythematous macules and/or papules | None |
| Drug hypersensitivity syndrome | Phenytoin, phenobarbital, carbamazepine, lamotrigine, allopurinol, sulfonamides, dapsone, minocycline, aspirin, vancomycin, azithromycin, abacavir, nevirapine, Chinese medicine | Edema (especially periorbital); erythematous macules and/or papules; sometimes vesicles or bullae | May be present |
| Lichenoid * | Captopril, enalapril, labetalol, nifedipine, propranolol, gold salts, hydrochlorothiazide, furosemide, spironolactone, hydroxychloroquine, ketoconazole, penicillamine, griseofulvin, tetracycline, carbamazepine, phenytoin, NSAIDs, hydroxyurea, imatinib, dapsone, sulfasalazine, allopurinol, iodides and radiocontrast media, IFN-γ, omeprazole, penicillamine, TNF inhibitors, sildenafil | Discrete flat-topped reddish-purple papules and plaques | May be present |
| Fixed drug | Sulfonamides, ibuprofen, acetaminophen, salicylates, tetracyclines, pseudoephedrine, loratadine, teicoplanin, metronidazole, macrolides, barbiturates, lamotrigine, potassium iodide, quinine, phenolphthalein, foods and food flavorings (especially tartrazine) | Solitary to few erythematous, hyperpigmented plaques | Unusual |
| Pustular (AGEP) | β-Lactam antibiotics, macrolides, clindamycin, terbinafine | Generalized small pustules and papules | None |
| Acneiform † | Corticosteroids, androgens, lithium, iodides, phenytoin, isoniazid | Follicular-based inflammatory papules and pustules predominate | None |
| Pseudoporphyria ‡ | NSAIDs, COX-2 inhibitors, tetracyclines, furosemide | Photodistributed blistering and skin fragility | None |
| Vasculitis § | Penicillins, NSAIDs, sulfonamides, cephalosporins | Purpuric papules, especially on the lower extremities; urticaria, hemorrhagic bullae, digital necrosis, pustules, ulcers | Rarely |
| Stevens–Johnson syndrome/toxic epidermal necrolysis | Sulfonamides, antiepileptics, NSAIDs, acetaminophen, allopurinol, dapsone | Target lesions, bullae, epidermal necrosis with detachment | Present |
| Drug-induced lupus ‖ | Minocycline, procainamide, hydralazine, isoniazid, penicillamine | Urticarial, vasculitic, erythematous | Rare |
* See Chapter 4 .
† See Chapter 8 .
‡ See Chapter 19 .
§ See Chapter 21 .
‖ See Chapter 22 .
Historical features
Drugs taken, date of initiation, and duration
Date of onset of eruption
Response to removal and rechallenge
Clinical features
Lesional morphology
Number of lesions and distribution
Involvement of mucous membranes
Associated features: pruritus, fever, lymphadenopathy, visceral involvement
Laboratory testing
Eosinophilia
Specific IgE levels
Prick and intradermal tests for late phase reactions (β-lactam allergy)
Provocation testing
IgE, Immunoglobulin E.
Allergic Reactions
Allergy may be defined as a specific acquired alteration in the capacity of an individual to react to an antigen. Mediated by circulating or cellular antibodies, allergic reactions may be classified as immunoglobulin (Ig) E-mediated hypersensitivity (type I), cytotoxic (type II), Arthus-type toxic immune complex reactions (type III), and delayed-type hypersensitivity (type IV). Type I / anaphylactic reactions include local and systemic manifestations of the interaction between antigen and tissue cells previously sensitized with skin-sensitizing reaginic antibody, usually IgE. This interaction of antigen and antibody results in the release of pharmacologically active substances that produce urticaria, angioedema, anaphylaxis, hay fever, and asthma. Type II / cytotoxic reactions include those reactions initiated by antibody interacting with an antigenic component of the cell surface. The antigen may be a natural component of the cell or an unrelated antigen that has become associated with the cell. Examples include pemphigus (see Chapter 13 ), hemolytic disease of the newborn, transfusion reactions, hemolytic anemia, leukopenia, or thrombocytopenia caused by the reaction of antibody with drugs attached to blood-cell surfaces. Type III / toxic immune complex reactions (Arthus-type reactions) are associated with the deposition of immune microprecipitates in or around blood vessels, which results in tissue damage through activation of complement or toxic products from leukocytes attracted to the areas. Examples include serum sickness, vasculitis, glomerulonephritis, and local Arthus reactions after injection of antigens into the skin, subcutaneous tissue, or muscle. Type IV / delayed-type hypersensitivity is the result of the interaction between antigen and specifically sensitized lymphocytic cells, which results in a mononuclear cell infiltration and the elaboration of toxic lymphoid cell products. Examples include tuberculin and other skin test reactions, allergic contact dermatitis (see Chapter 3 ), lichenoid drug eruptions (see Fig. 20-1 and Chapter 4 ), fixed drug eruptions, TEN, and graft-versus-host-type reactions (see Chapter 25 ). Patch testing may be useful in determining the cause of allergic contact dermatitis (see Chapter 3 ).
Urticaria
Urticaria, a systemic disease with cutaneous manifestations, occurs at some time in the life of about 15% to 20% of all persons. It is characterized by the appearance of transient well-circumscribed skin lesions that present as erythematous, intensely pruritic elevated swellings (wheals) of the skin or mucous membranes. More than 80% of cases of new-onset urticaria resolve in 2 weeks and more than 95% of new-onset cases within 3 months.
Urticaria usually represents a type I (or immediate) hypersensitivity reaction in which the protein or its metabolite binds to IgE on the surface of cutaneous mast cells, leading to activation, degranulation, and release of vasoactive mediators such as histamine, leukotrienes, and prostaglandins. Examples of immunologically mediated urticaria are reactions to latex and reactions to peanuts (see below). Urticaria may also result from nonimmunologic triggering of mast cell release, such as from radiocontrast media, aspirin, many types of nonsteroidal anti-inflammatory drugs (NSAIDs), or opiates.
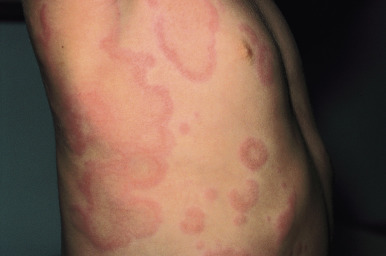
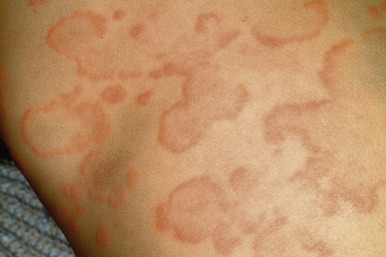
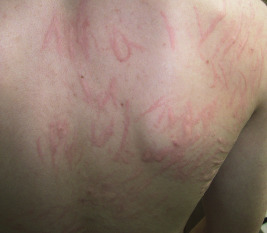
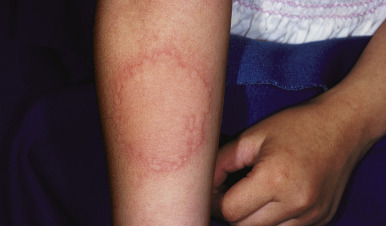
Typical lesions of urticaria have an edematous center with a halo of erythema ( Fig. 20-2 ). They vary from pinpoint-sized papules to large lesions several centimeters in diameter. Central clearing, peripheral extension, and coalescence of individual lesions result in a clinical picture of oval, annular, or sometimes bizarre serpiginous configurations ( Figs. 20-3 and 20-4 ). When annular or serpiginous patterns of urticaria occur, they can be confused with erythema multiforme (EM) or serum sickness reactions ( Fig. 20-5 ), and the designation urticaria multiforme has been proposed. Other mimics of urticaria include urticarial vasculitis, Henoch–Schönlein purpura, acute hemorrhagic edema of infancy (see Chapter 21 ), systemic onset juvenile idiopathic arthritis (see Chapter 22 ), and cryopyrin-associated periodic syndromes (see Chapter 25 ).
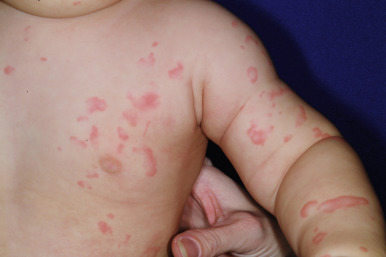
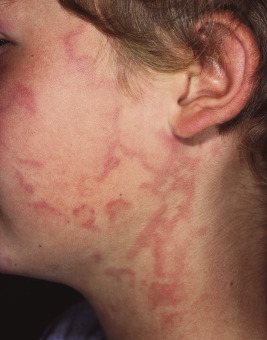
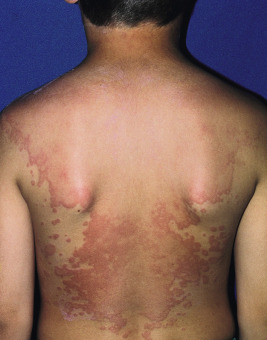
Urticaria may be localized to one small area or may become generalized ( Fig. 20-6 ). Subcutaneous extension may result in giant wheals. In infants and young children, swelling of the distal extremities with acrocyanosis may be a prominent feature of the urticarial reaction. Occasionally, particularly in infants and young children, bullae may form in the center of the wheal, usually on the legs and buttocks. Individual wheals rarely persist longer than 12 to 24 hours. Parents can draw a circle around a few lesions to verify clearance of each individual lesion by approximately 24 hours or less. Those lasting longer than 24 to 36 hours are probably not true urticaria and may represent another vascular pattern such as urticarial vasculitis (see Chapter 21 ) or EM.
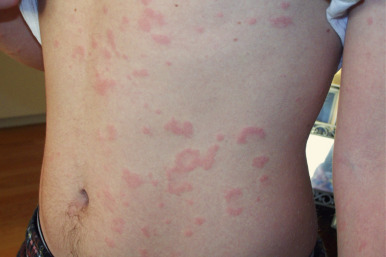
Urticaria of less than 6 weeks’ duration is considered acute. In 80% of children, acute urticaria is the result of infection, often viral, whereas in adolescents food and drugs play a more significant role. Among foods, dairy products play a more common role in younger children, in contrast to nuts, seafood, berries, and grains in older children. Streptococcal infection, mycoplasma, histoplasmosis, and coccidiomycosis in endemic areas are also well-known triggers, although almost every infectious agent has been associated with urticaria. Overall, up to 10% of episodes of urticaria are related to drugs, and these are usually acute in nature. The penicillins are the most common drugs to trigger urticaria, but only 10% to 20% of individuals who report allergy to penicillin are really allergic when skin prick testing to penicilloyl-poly-lysine and then oral drug challenge (if negative) is performed. Other drugs most likely to induce an urticarial drug reaction are cephalosporins, sulfonamides, tetracyclines (especially minocycline), antiepileptics, and monoclonal antibodies, including omalizumab. Although cross-intolerant reactions are more common than IgE-mediated reactions with NSAIDs, selective immediate reactions to NSAIDs are becoming increasingly recognized with the widespread use of these medications in children (especially ibuprofen). In vitro testing (immunoassays and basophil activation testing) can detect specific IgE antibodies for a limited number of drugs and complement prick testing but should be performed within 1 year after the occurrence of a reaction. Anaphylactoid reactions (anaphylaxis without identifiable specific IgE antibodies) most commonly result from ingestion of acetylsalicylic acid. Non-immunologically mediated urticaria may result from radiocontrast media and NSAIDs. Psychologic stress may exacerbate (but not cause) urticaria.
Urticaria that recurs often and lasts longer than 6 weeks is termed chronic. Chronic urticaria is much more common in adults than in children ; it may be present continuously or nearly every day (“chronic continuous urticaria”) or as attacks separated by symptom-free periods of up to several weeks (“chronic recurrent or intermittent urticaria”). Diagnosis and treatment of a patient with chronic urticaria demands a complete history and physical examination, appropriate laboratory evaluation, and an awareness of possible underlying disease.
If the trigger is not clear, a child may have idiopathic urticaria. However, 30% to 47% of children with chronic urticaria are now recognized to have autoimmune (also called autoreactive ) urticaria. Autoimmune urticaria is caused by IgG antibodies recognizing the high-affinity IgE receptor (FcεRIα) or less commonly, IgE itself. Patients may have autoimmune thyroid disease and less commonly, celiac disease, type 1 diabetes, juvenile idiopathic arthritis, and systemic lupus erythematosus ( Fig. 20-7 ). Reactions from food or food additives have been described in 9%. However, the urticaria from food additives, artificial flavoring, or preservatives is not IgE-mediated but rather is a pseudoallergic reaction that is often delayed (4 to 12 hours after ingestion). Among less common possible triggers of chronic urticaria are physical causes, foods and food additives, drugs (especially aspirin and NSAIDs), immunizations, insect bites, inhalant or contact allergens, and infections (parasites, Helicobacter pylori , Chlamydia, dental abscesses, sinus infections, low-grade urinary tract infection, and otitis). Chronic urticarial lesions with high fevers may also suggest cryopyrin-mediated auto-inflammatory disease (see Chapter 25 ). Urticarial vasculitis may be confused with chronic urticaria, but lesions persist beyond 24 hours (see Chapter 21 ).
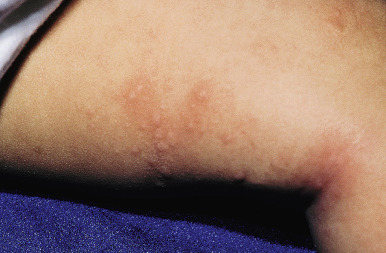
In pediatric studies, remission rates at 1, 3, and 5 years after the onset of chronic urticaria range from 16% to 18%, 39% to 54%, and 50% to 68%, respectively, regardless of whether the chronic urticaria is autoimmune, although being female and older than 10 years of age are poorer prognostic signs.
Further investigation should be based on the known underlying causes, history, and examination and could include complete blood count, erythrocyte sedimentation rate, antinuclear antibody, CH50, free-T4, thyroid stimulating hormone, antithyroglobulin and antimicrosomal antibodies, skin prick tests for foods, food challenges, and stool examination for parasites. Food avoidance correlates better with a positive history of food reactivity than with positive prick testing. The autologous serum skin test (ASST) through intradermal injection of the patient’s own serum is used to confirm autoreactivity and is more sensitive than the in vitro histamine-release urticaria test. However, new enzyme-linked immunosorbent assay (ELISA) and immunodot tests have recently been developed to measure anti-FcεRIα antibodies directly.
Effective treatment of urticaria depends on identification of the etiologic factor and its elimination whenever possible. First-line treatment in children is administration of a nonsedating histamine (H1) blocker such as cetirizine, loratadine, desloratadine, fexofenadine, or levocetirizine. Dose escalation may then be required up to twofold to fourfold those used for management of allergic rhinitis. Addition of hydroxyzine, diphenhydramine, or another sedating H1 blocker at night may be considered but should not replace nonsedating antihistamines. Sedating antihistamines are best administered about 1 hour before bedtime to minimize daytime drowsiness. Paradoxical hyperactivity and irritability, which requires discontinuation of the medication, has occasionally been described in some infants and children with the administration of H1 blockers, especially diphenhydramine. Doxepin, a tricyclic antidepressant with H1- and H2-antagonistic effects, has also been utilized for the treatment of chronic urticaria but may lead to intolerable sleepiness. Approximately 80% of patients respond to antihistamines. Concomitant administration of an H2 blocker (such as ranitidine or cimetidine) tends to be of limited value, but a leukotriene receptor antagonist (e.g., montelukast) can be considered if antihistamines are insufficient. Antihistamine administration should be continued for a few weeks after clearance of the urticaria with gradual tapering of the dosage in order to determine whether they are still needed for control. Avoidance of aspirin and NSAIDs may be helpful, because these medications may exacerbate chronic urticaria. Monthly administration of subcutaneous omalizumab (150 to 300 mg for adolescents and adults) or cyclosporine may be required for chronic urticaria. Dapsone, methotrexate, mycophenolate mofetil, cyclophosphamide, hydroxychloroquine, sulfasalazine, intravenous immunoglobulin (IVIG), and plasmapheresis have all been used for resistant chronic urticaria but have less evidence than omalizumab and cyclosporine. The subcutaneous administration of 0.1 to 0.5 mL of epinephrine 1 : 1000 is often effective for patients with angioedema and severe urticaria. Although often effective in patients with severe or persistent urticaria, administration of systemic corticosteroids should be reserved for acute reactivity in recalcitrant patients and should be limited to 10 days because of the many potential side effects.
Contact Urticaria
Urticaria may also occur from skin exposure to antigen rather than through ingestion or inhalation ( Fig. 20-8 ). In this case, the lesions tend to be localized to the site of contact, although rarely satellite lesions or even more generalized involvement has occurred. Exposure to certain jellyfish, corals, caterpillars, moths, and chemicals can lead to contact urticaria through pharmacologic means. Immunologic reactions can also result in contact urticaria, as often occurs with exposure to fish or latex. Localized small wheals have also been described after contact with the spines of pet hedgehogs. Aquagenic urticaria is a variant of contact urticaria in which small urticarial papules arise at follicles after exposure to water.
Physical Urticarias
The physical urticarias are a group of disorders in which wheals occur in response to various physical stimuli. These include dermographism and pressure, cholinergic, aquagenic, solar (see Chapter 19 ), and cold urticaria. Dermographism and cholinergic urticaria are quite common; cold urticaria is less common; and other patterns of physical urticaria are relatively rare.
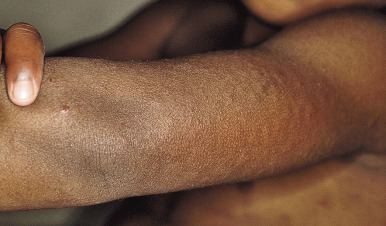
Dermographism (dermatographism) is manifested by a sharply localized edematous or wheal reaction with a surrounding zone of erythema that occurs precisely at the site and within seconds of firm stroking of the skin ( Fig. 20-9 ). The wheal tends to be maximal in size and intensity at about 6 minutes after onset and persists for approximately 15 minutes. This common phenomenon, known as the triple response of Lewis, is often seen in infants, occurs in about 50% of children, and is noted in only about 1% of adolescents or adults.
Pressure urticaria is a variant of dermographism characterized by the development of hives or a deeper swelling simulating angioedema after local pressure such as from clothing or jewelry, or weight bearing. The reaction is often painful rather than pruritic and may occur immediately after the pressure or more commonly, after a 4- to 6-hour delay (“delayed pressure urticaria”). Because of this delay in appearance, patients often fail to appreciate the cause of the disorder. The palms and soles are most commonly involved. Patients respond poorly to antihistamine therapy; some patients have responded to the leukotriene antagonist, montelukast.
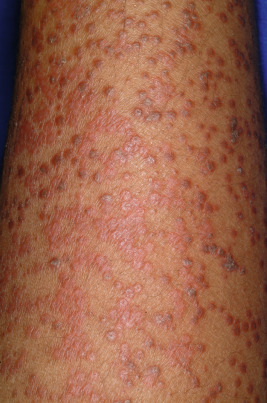
Cholinergic urticaria (micropapular urticaria), which occurs in 5% to 7% of individuals with urticaria, is a very distinctive type. It usually starts in adolescence and is associated with heat, exertion, or emotional stress. Cholinergic urticaria is characterized by a generalized eruption, especially on the trunk and arms, which consists of discrete, papular wheals, 1 to 3 mm in diameter, with or without a surrounding area of erythema ( Fig. 20-10 ). The wheals first appear within minutes after sweating begins and occasionally are accompanied by exercise-induced bronchospasm, headache, gastrointestinal discomfort, and faintness. The duration of the eruption varies from 30 minutes to several hours; after an episode, a refractory period of about 24 hours ensues. A subset of patients with cholinergic urticaria have hypohidrosis or anhidrosis.
Acetylcholine is thought to participate in the pathomechanism of wheal formation, but immediate type skin responses to one’s own sweat, and less commonly, positive ASSTs have been described. Patients who react to their own sweat usually show nonfollicular wheals with satellites, whereas those individuals with cholinergic urticaria and positive ASST testing show a follicular-based process without satellite wheals. Once cholinergic urticaria occurs, the condition may recur for periods of months to years and then tends toward spontaneous improvement and resolution. Treatment consists of systemic antihistamines, particularly cyproheptadine, hydroxyzine, and nonsedating antihistamines such as cetirizine; awareness of potential precipitating factors; and avoidance of heat, excessive exertion, and excitement whenever possible. Another option is the use of oral anticholinergic medications to decrease sweating.
Aquagenic urticaria , a disorder that resembles cholinergic urticaria, occurs most often in adolescence and is characterized by small, intensely pruritic, perifollicular papular wheals with surrounding erythema ( Fig. 20-11 ). The palms and soles are spared. The disorder is precipitated by contact with water or perspiration (irrespective of temperature). Exercise and other cholinergic factors do not precipitate this disorder. Patients can drink water without adverse reaction. Antihistamines often improve reactivity, but do not totally suppress it.
Cold urticaria accounts for approximately 3% of chronic urticaria and has been documented in infants as young as 6 months. It is characterized by localized or generalized urticaria, sometimes with angioedema or anaphylaxis, that develops within a few minutes or hours after exposure to cold air or water, often upon rewarming. The urticaria may be confined to the area of skin in contact with cold (in milder cases) but may be generalized. In highly sensitive individuals, it may be associated with respiratory or cardiovascular compromise, especially with water immersion. The result is hypotension and on occasion, syncope, loss of consciousness, and drowning. Respiratory signs such as nasal stuffiness, cough, and dyspnea, and oral or gastrointestinal symptoms such as swelling of the lips, swelling of the oral mucous membranes, dysphagia, and abdominal cramps may occur. Patients who experience oropharyngeal reactions to cool liquids or foods (especially lip swelling) are at increased risk for the development of systemic reactions.
Cold urticaria in children and adolescents is usually acquired, occurs more often in girls, and often appears suddenly. Anaphylaxis has been described in up to 30% at presentation. Once symptoms develop, they are generally short-lived and recurrences usually disappear after a few months or years. Secondary forms of cold urticaria may also be associated with cold hemolysin and cold agglutinin syndromes. These forms, generally seen in adults, cause Raynaud phenomenon, acrocyanosis, and cutaneous ulcers. Some cases of cold urticaria manifested by itching, erythema, purpura, atypical Raynaud phenomenon, and ulceration are the result of cryoglobulins (see Chapter 21 ).
Cold urticaria can rarely be a manifestation of familial cold auto-inflammatory syndrome (FCAS), an autosomal dominant disorder resulting from mutations in CIAS1/NLRP3 (see Chapter 25 ). It is characterized by an urticarial or papular eruption, fever, chills, arthralgia, and sometimes headache, malaise, muscle tenderness, and significant leukocytosis. Patients more commonly complain of burning or stinging than of pruritus. Although the tendency to familial cold urticaria generally persists for life, the severity may decrease with advancing age. It generally develops after a latent period of several hours, and once it develops, it may persist for up to 48 hours. Familial atypical cold urticaria (FACU) is an autosomal dominant disorder with onset during childhood in which the urticaria and/or angioedema develop shortly after cold exposure (not delayed). Cold-stimulation testing is negative (no wheal), and in contrast with FCAS, patients do not have fever, chills or joint complaints. Phospholipase C, gamma 2 (PLCG2)-associated antibody deficiency and immune dysregulation (PLAID) is a newly characterized autosomal dominant immunodeficiency syndrome associated with evaporative cold urticaria. The cold urticaria presents within the first year in all patients and is characterized as pruritic blotchy macules but not true wheals; ice cube testing is negative. Patients may develop ulcerative lesions of the nasal tip, and occasionally fingers and toes, during the neonatal period that heals by infancy. They have a tendency to develop granulomatous dermatitis, which may be widespread. Recurrent sinopulmonary infections, common variable immunodeficiency (CVID), and autoantibodies (especially antinuclear and anti-thyroid antibodies) are commonly associated.
The diagnosis of cold urticaria requires a careful history and investigation for other possible etiologic factors. The diagnosis may be confirmed by reproducing signs through local application of an ice cube for periods of 2 to 10 minutes with observation after removal for at least 10 minutes. The best areas for this testing are the face, neck, and particularly the arms ( Fig. 20-12 ). Some patients fail to respond to ice but do respond to cold water or generalized cooling of the body. Critical temperature threshold testing shows an inverse relationship with disease severity and activity, which can be helpful for assessing both the severity and the impact of therapy. Cold urticaria should be distinguished from cold-induced cholinergic urticaria, in which cholinergic urticaria occurs while exercising in the cold and is not induced by ice cube testing. Immediate ice cube tests also tend to be negative with FCAS, FACU, and PLAID syndromes.
Underlying disorders are rare in children. Cryoglobulinemia, cryofibrinogenemia, and cold agglutinins have all been reported, particularly in adults, and can reflect an underlying diagnosis of essential mixed cryoglobulinemia, hepatitis, autoimmune disease, or lymphoma. Cold urticaria can also be associated with infectious disorders (toxoplasmosis, Epstein–Barr virus [EBV], H. pylori, hepatitis C, and human immunodeficiency virus [HIV]) and autoimmune disease (particularly lupus erythematosus or celiac disease).
Patients with cold urticaria, and particularly with severe or widespread urticarial reactions, should carry epinephrine and be forewarned of the risk of drowning after loss of consciousness when swimming or bathing in cold water. The treatment of cold urticaria is aided by oral administration of antihistamines, particularly nonsedating H1 blockers, although the sedating antihistamine cyproheptadine is also particularly helpful for cold urticaria, and leukotriene receptor antagonists have been used adjunctively. Increases in doses of nonsedating antihistamine to four times the standard dosages have shown additional benefit (vs. standard dosing) without increased adverse events. For those patients who are unresponsive to systemic antihistamines and have increased levels of IgE, treatment with omalizumab may be helpful. Desensitization to cold is an alternative that is now rarely performed; an extremity is gradually cooled in cold water for 5 to 10 minutes per day with a gradual increase in the time of exposure and decrease of the temperature over a period of weeks or months. This treatment is not regularly effective and must be done cautiously in an effort to minimize the risk of systemic reaction. Patients with cold urticaria as a manifestation of an auto-inflammatory syndrome often require interleukin (IL)-1 blockers (e.g., anakinra) for response.
Anaphylaxis
Anaphylaxis is a life-threatening, immediate hypersensitivity reaction to the administration of an antigen that has previously produced a specific sensitization. It is characterized within minutes to 1 hour after injection or ingestion of antigen by pruritus of the palms, soles, and scalp, urticaria, and/or angioedema with systemic signs (weakness, dyspnea, hypotension, and circulatory collapse). Mucosal involvement of the airway leads to acute respiratory distress, and swelling of the gastrointestinal mucosa can result in abdominal pain, vomiting, or diarrhea. The incidence in children and adolescents is 10.5 episodes per 100,000 person-years, and most children have a personal history of atopy. Anaphylaxis can occur during infancy, although the risk peaks during early childhood and decreases thereafter. A biphasic reaction may occur, with recurrence usually within 8 hours of the initial episode. Anaphylactic reactions most often occur after ingestion of foods, especially peanuts, cow’s milk, and hen’s eggs in children (vs. wheat and shellfish in adults), or in response to drugs or insect venom. Anaphylactic reactions to latex and peanuts are particularly troublesome, because they are so ubiquitous in the environment. Having asthma as well as IgE-mediated food allergy increases the risk of developing food-induced anaphylaxis. Exercise-induced anaphylaxis may also occur and is commonly food-dependent. Two subtypes have been described: nonspecific food-dependent exercise-induced anaphylaxis, in which filling the stomach before exercise is responsible, regardless of the kind of food ingested, and specific food-dependent exercise-induced anaphylaxis, an IgE-mediated food allergy in which anaphylaxis only occurs in combination with exercise. Several types of foods have been described to trigger exercise-induced anaphylaxis, but wheat appears to be most common.
Latex Allergy
Latex allergy is a reaction to a peptide in natural rubber and occurs most commonly in children with spina bifida, presumably owing to the frequent exposure to catheters and latex gloves. The use of latex-free or latex-safe equipment has markedly lowered the risk to children with spina bifida. Atopic children also show an increased risk of latex allergy, with up to 4% showing prick test reactivity. Type I hypersensitivity reactions to latex may range from contact urticaria to life-threatening anaphylaxis. Severe anaphylactic shock without cutaneous erythema or urticaria has been described. Type I reactions to latex differ from the type IV contact dermatitis reaction to antigens in rubber products (see Chapter 3 ). Children with type I hypersensitivity reactions often experience lip swelling after sucking on a pacifier or blowing up a balloon ( Table 20-2 ). Aerosolized powder from latex gloves and inhalation may lead to runny nose, sneezing, itchy, swollen eyes, and wheezing in susceptible individuals. Approximately half of persons who react to latex show reactivity to certain foods, resulting in oral mucosal pruritus, urticaria, and wheezing; bananas, kiwis, avocados, and chestnuts contain proteins that cross-react with latex and are most commonly associated.
| Household | Medical |
|---|---|
| Balloons | Adhesive tape |
| Balls (basketball, Koosh, squash, tennis) | Bandages |
| Cell phone cases | Blood pressure cuffs |
| Computer mouse pads | Bulb syringes |
| Condoms | Catheters |
| Disposable diapers/rubber pants | Dental devices |
| Erasers | Electrode pads |
| Gloves for dishwashing | Face masks and their straps |
| Pacifiers, nipples | Gloves |
| Rubber bands | Injection ports |
| Rubber cement | Rubber syringe stoppers |
| Rubber raincoats and rubber boots | Stethoscope tubing |
| Shoe soles | Tourniquets |
| Socks with elastic | Vial stoppers |
| Sports equipment | Wound drains |
| Swimming masks and goggles | |
| Toys | |
| Underwear |
Skin prick testing is most likely to yield positive responses to either latex or cross-reacting foods. Serum latex-specific IgE by Immuno CAP can also be used to detect type I reactivity to latex, but the high rate of false positives should be taken into account when making a diagnosis of latex allergy in patients with pollen allergy (30%), especially in those sensitized to more than one pollen species. Avoidance of latex and related triggers is critical, and nonlatex substitutes such as Mylar balloons and vinyl or neoprene gloves are generally available. Desensitization is the only effective way to resolve latex allergy; percutaneous and sublingual approaches appear to be safer than subcutaneous administration.
Nut Allergy
Nut-induced anaphylaxis is a risk in the 1.1% of individuals who are allergic to peanuts and tree nuts and has resulted in 50 to 100 deaths annually in the United States. Screening tests such as prick tests and radioallergosorbent tests (RASTs) are suggestive, but only food challenge confirms reactivity. Avoidance of peanuts is extremely difficult, and there has been no means to protect against accidental ingestion, which often occurs despite reassurance from servers. Allergic reactions through kissing are well documented, particularly in adolescents. In one study, 13% of subjects still had detectable levels of peanut 1 hour after ingestion of two tablespoons of peanut butter on a sandwich. Although prenatal and postnatal avoidance of peanuts has been encouraged for at-risk individuals in the past, new data suggests that exposure to small amounts can lead to tolerance. In a landmark trial of 500 infants at risk for peanut allergy, infants who avoided peanuts had a much higher risk of developing allergy than those who were given small numbers of peanuts regularly, regardless of whether they were sensitized to peanuts at baseline. These data have led to the suggestion that 4- to 8-month-old infants at risk for peanut allergy with negative prick testing for peanuts or mildly positive prick testing and a nonreactive peanut challenge should be administered 2 g peanut protein three times weekly for at least 3 years.
Omalizumab substantially increases the threshold of sensitivity to peanuts and may protect against unintended ingestion. Oral immunotherapy with gradual increases in exposure to peanuts can decrease basophil activation, titrated skin prick tests, and peanut-specific IgE levels within 6 months.
The treatment of anaphylaxis consists of the intravenous administration of antihistamines such as diphenhydramine (1.25 mg/kg), subcutaneous epinephrine (the adult dose AutoInjector contains 0.3 mg of epinephrine as 0.3 mL of epinephrine 1 : 1000, and the pediatric dose AutoInjector contains 0.15 mg of epinephrine as 0.3 mL of epinephrine 1 : 2000) for grade 2 anaphylaxis or higher, and sometimes intravenous corticosteroid. Epinephrine is the only medication shown to be life-saving when administered promptly, but it is underutilized. Patients should have easy access to at least 2 doses of an epinephrine AutoInjector, with thorough training regarding correct use of a given device and an emergency action plan. In addition to EpiPen, Auvi-Q is a new AutoInjector available for children (and adults) that provides audio and visual cues regarding patient use and was found favorable to the traditional AutoInjector for method of instruction, preference to carry, and device size.
Because of the delay before onset of effectiveness, oral medications are not indicated. If hypotension is present, intravenous fluids should be initiated to permit the rapid administration of plasma volume expanders, fluids, and electrolytes. Once shock is overcome and oral fluids are well tolerated, oral corticosteroids may be initiated. Anyone at risk for anaphylaxis should wear a medic alert bracelet and carry epinephrine in case of emergency. Management of anaphylaxis in schools has recently received attention, especially because of its distinct challenges and the need to be able to urgently administer epinephrine.
Angioedema
The term angioedema (also called giant urticaria or Quincke edema ) describes swelling of the eyelids, hands, feet, genitalia, the lips, tongue, airway, and gastrointestinal tract. When it occurs in association with wheals, angioedema is a feature of urticaria. Fifty percent of patients with angioedema also show urticaria, and 10% of infants and children with urticaria show at least mild angioedema. Angioedema may be a component of anaphylactic reactions.
Angioedema without wheals has been recently classified by the European Academy of Allergy and Clinical Immunology into four acquired and three hereditary forms. Angioedema is defined as hereditary when: (1) there is family history of angioedema in a first- or second-degree relative; (2) there is a mutation in SERPING1 (encoding C1 inhibitor [C1-INH]) or FXII (encoding factor XII, a coagulation factor); and (3) there is a familial deficiency of C1-INH. Although factor XII deficiency is more uncommon than C1-INH-hereditary angioneurotic edema (HAE), the clinical features of deficiency of C1-INH and deficiency of factor XII are similar, and laboratory testing is required to distinguish these autosomal dominant disorders. Two of the three acquired forms (induced by angiotensin-converting enzyme inhibitors and a nongenetic form of C1-INH deficiency) usually occur in adults. Most cases of idiopathic acquired angioedema are histamine-responsive (histaminergic; termed idiopathic histaminergic acquired angioedema [ IH-AAE ]), and medications (especially aspirin and NSAIDs), allergens, and physical agents have been implicated.
C1 Inhibitor-Hereditary Angioneurotic Edema
Hereditary angioneurotic edema (HAE; hereditary angioedema) is characterized by recurrent episodes of edema of the subcutaneous tissue, particularly the hands, feet, and face, and of the gastrointestinal and/or upper respiratory tracts. Occurring in approximately 1 in 50,000 to 1 in 10,000 persons, the defect results from a deficiency (85% of patients; type I) or dysfunction (15% of patients, type II) of the inhibitor of the first component of complement [C1-INH]). Deficiency or dysfunction of C1-INH leads to transient episodes of increased vascular permeability with increased cleavage of C4 by C1, depleting C4 levels. The major factor responsible for edema formation is now known to be bradykinin, which can induce leakage of postcapillary venules. Bradykinin is produced when high molecular-weight kininogen is cleaved by plasma kallikrein, the activity of which is controlled by C1-INH. HAE without urticaria and with no dysfunction or deficiency of C1-INH has been described.
The earliest symptoms of HAE often begin in infancy or early childhood, with 50% of cases manifesting before the age of 10 years. The frequency and severity of attacks are typically exacerbated during adolescence and subside in the fifth decade. Onset during the first decade is associated with greater severity. Based on survey data, prodromes have been described in 82.5% to 95.7% of patients surveyed, most commonly involving the skin or gastrointestinal tract and hours to days before the onset of angioedema; fewer than 10% reported were rarely or never able to predict an attack. Affected individuals are prone to sudden attacks of circumscribed subcutaneous edema (angioedema) without associated urticaria. It usually affects the face or an extremity and may be severe enough to cause remarkable disfiguration of the affected parts. Areas of swelling may migrate, but the condition generally subsides within 24 to 72 hours. Several weeks of remission generally follow attacks. The skin and mucosal lesions may appear spontaneously or may be precipitated by minor trauma, especially dental work or surgery, strenuous exercise, infection (especially of the airway), menses, pregnancy, administration of oral contraceptives or angiotensin-converting enzyme inhibitors, extremes of temperature, or emotional disturbances. There is no pitting, discoloration, redness, pain, or itching associated with the edema. However, many affected children show a nonpruritic serpiginous rash that resembles erythema marginatum.
Gastrointestinal involvement is the presenting sign in 40% to 80% of affected children. The recurrent colic and severe abdominal pain can simulate the acute abdominal pain of appendicitis. Nausea, vomiting, and diarrhea are additional complaints. Abdominal ultrasound may demonstrate edematous swelling of the intestinal wall and free peritoneal fluid. Involvement of the mucous membranes of the hypopharynx and larynx, although seen less often, may be particularly devastating; up to 25% of patients experience asphyxia, which is the leading cause of death and usually occurs during the third decade of life.
Diagnosis of HAE can be confirmed by the finding (at least twice and separated by 1 to 3 months) of reduced levels of C4 or C1-INH (quantitative and/or functional), or both. For those with C1-INH deficiency, levels tend to be 10% to 40% of normal, reflecting the haploinsufficiency. Of note, C4 and C1-INH levels do not tend to reach adult levels until about 3 years of age, necessitating genetic testing in infants. If levels of C4 are reduced in the presence of normal levels of C1-INH protein, confirmation must rely on demonstration of a functional lack of C1-INH by chromogenic or immunoenzymatic assay.
Antihistamines and corticosteroids have not been effective in the management of patients. Epinephrine is beneficial in the control of swelling in only a very few patients. Tracheostomy commonly is lifesaving in patients with laryngeal obstruction, but plasma-derived human C1-INH concentrate is available for treatment of acute episodes. The median time to onset of relief in one pediatric study was 25 minutes and to complete resolution was 8 hours. The US Hereditary Angioedema Association and others have proposed recommendations for comprehensive care of patients with HAE. Recombinant C1-INH from transgenic rabbits has recently been approved for use in acute attacks. Ecallantide, a specific inhibitor of plasma kallikrein, and icatibant, an inhibitor of the bradykinin receptor B2, are both used for acute attacks, but they have short half-lives and are not appropriate for prophylaxis.
In general, attacks tend to be less frequent and less severe in children, and most will not require long-term prophylaxis. However, prophylactic C1-INH administration with dosage escalation may be considered for children with more than two attacks per month, the need for acute treatment more than once annually, or more than one episode of severe abdominal pain in a year. When used prophylactically, the concentrate reduced the median monthly attack rate by almost eightfold. Antifibrinolytics (ε-aminocaproic acid and tranexamic acid) are less efficacious than other approaches and are now rarely used for prophylaxis. Attenuated androgens such as danazol are only appropriate for short-term prophylactic therapy, such as before dental procedures, because of their potential side effects.
As prevention, children and adolescents with HAE should not participate in contact sports, and toddlers should not attend daycare before kindergarten to minimize the risk of infection that could trigger attacks. Even the eruption of teeth and minor mechanical trauma can induce edema formation. If menstruation worsens HAE symptoms, long-term prophylactic doses can be increased, but oral contraceptives are triggers of the angioedema and should be avoided. Although use of angiotensin-converting enzyme inhibitor is unusual in children, these medications can exacerbate the signs of angioedema in affected individuals and should be avoided.
Scombroid and Ciguatera Fish Poisoning
Scombroid fish poisoning (scombrotoxism) is a clinical syndrome that results from the ingestion of spoiled fish of the Scombroidea family (tuna, mackerel, and bonito) and fish such as bluefish, mahi-mahi, amberjack, herring, sardines, and anchovies. The disorder, thought to be related to high levels of histamine and saurine produced when these fish are improperly refrigerated, is characterized by pruritus, a diffuse erythema of the face and upper body, somewhat resembling a sunburn and at times, giant hive-like lesions that develop within minutes to hours after ingestion of the toxic fish. Bacteria in the fish express a decarboxylase that converts histidine to histamine, which resists cooking and freezing. Symptoms resemble a histamine reaction and commonly include a hot, burning sensation rather than pruritus. The conjunctivae are often markedly injected, and many patients have a severe throbbing headache, tachycardia, palpitations, nausea, vomiting, abdominal cramps, diarrhea, dryness and a burning sensation or peppery taste in the mouth, urticaria, angioneurotic edema, oral blistering, hypotension, blurred vision, and asthma-like symptoms. Although superficially resembling an allergic reaction, patients with scombroid fish poisoning can be reassured that they do not have fish allergy and that scombroidosis will not occur when fish are handled properly. Symptoms are generally self-limiting and even if untreated tend to resolve within 8 to 10 hours. Treatment is supportive, and most patients treated with oral antihistamines become asymptomatic within 2 or 3 hours.
In contrast to scombroid fish poisoning, ciguatera fish poisoning is a common disorder endemic throughout the Caribbean and Indo-Pacific islands but has been reported in the United States. It is caused by the ingestion of ciguatoxin, which originates from a dinoflagellate ( Gambierdiscus toxicus ). Present in certain fish such as the red snapper, amberjack, and sturgeon, ingestion of affected fish (whether cooked, raw, or frozen) may result in this disorder. Clinical symptoms, which usually appear within 12 hours but sometimes within minutes after ingestion of ciguatoxin, include abdominal pain, cramping, diarrhea, nausea, vomiting, paresthesias, pain or burning when cold water is touched, arthralgia, myalgia, and in severe cases, hypotension, shock, respiratory depression, paralysis, coma, and death. The duration of illness averages 8.5 days but may be prolonged. Treatment consists of supportive measures, and intravenous mannitol has been found to be extremely effective, lessening the neurologic and muscular dysfunction of affected patients within minutes of administration.
Serum Sickness and Serum Sickness-Like Reactions
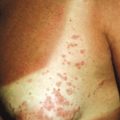
Serum sickness is an allergic reaction characterized by a cutaneous eruption (morbilliform, urticarial, or purpuric), malaise, fever, lymphadenopathy splenomegaly, proteinuria, and arthralgias. The syndrome, originally noted and most commonly seen after the administration of antiserum of horse or rabbit origin, is now rarely encountered. However, serum sickness-like reactions (SSLRs) occasionally occur in children, especially 1 to 3 weeks after exposure to cefaclor, which has been described in approximately 0.2% of treated children. Penicillins (especially amoxicillin), tetracyclines, cefprozil, sulfonamides, macrolides, ciprofloxacin, rifampin, griseofulvin, itraconazole, fluoxetine, bupropion, rituximab, and the H1N1 influenza vaccine have also been implicated as triggers of SSLR. SSLR more closely resembles urticaria (or urticaria multiforme) than true serum sickness ( Figs. 20-13 and 20-14 ), and lesions commonly show large areas of lilac or violaceous discoloration, especially centrally ( Fig. 20-15 ). This classic observation has led to use of the term purple urticaria to describe the skin lesions seen in SSLR. Noncutaneous features most commonly include fever, malaise, lymphadenopathy, and arthralgias with periarticular swelling, particularly symmetrically involving the knees and metacarpophalangeal joints ( Fig. 20-16 ). True arthritis is uncommon and seen significantly less often than in patients with true serum sickness. Affected children may also demonstrate facial edema, eosinophilia, headache, myalgia, and gastrointestinal symptoms, but renal and neurologic disease is rarely associated. Histologic evaluation of biopsy section may reveal vasculitis associated with cutaneous deposition of immune complexes, but hypocomplementemia and circulating immune complexes, as seen in true serum sickness, are absent.
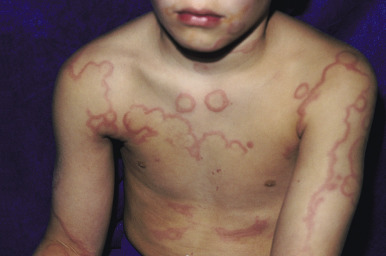
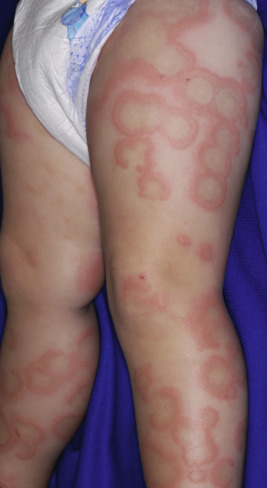
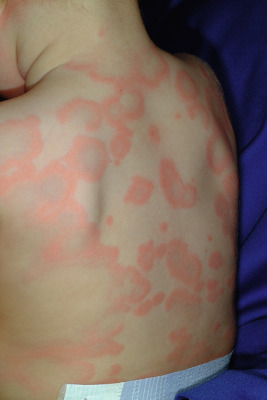
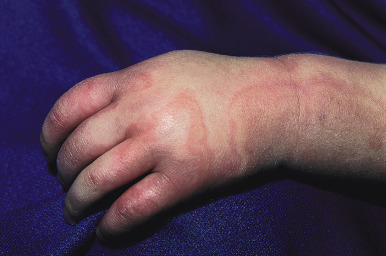
SSLR generally is a self-limiting disease that subsides within 2 to 3 weeks after discontinuation of the causative agent. In the case of cefaclor, biotransformation of the parent drug, genetic defects in metabolism of reactive intermediates, and increased in vitro lymphocyte cytotoxicity have been thought to participate. SSLR has been described as well in association with other drugs, infection with hepatitis B or C, and immunization against hepatitis B, rabies, and tetanus toxoid. Treatment with antihistamines, NSAIDs, and if severe, a short course of systemic corticosteroids helps to alleviate symptoms. In general, the risk of cross-reaction among β-lactam antibiotics is low, and patients who have reacted to cefaclor or cefprozil will usually tolerate other cephalosporins.
Exanthematous Eruptions
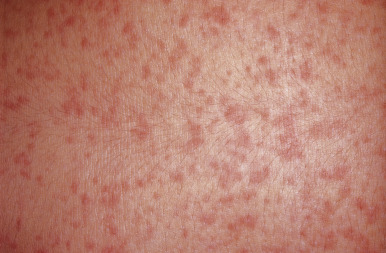
The most common drug eruptions are morbilliform or exanthematous eruptions, characterized by erythematous macules and/or papules ( Figs. 20-17 and 20-18 ). The risk of this type of eruption is increased by viral infection, such as the near 100% incidence of an exanthematous reaction in patients taking penicillin who have EBV infection, or the increased risk of drug reactions in patients taking sulfonamides who have human immunodeficiency virus (HIV) infection.
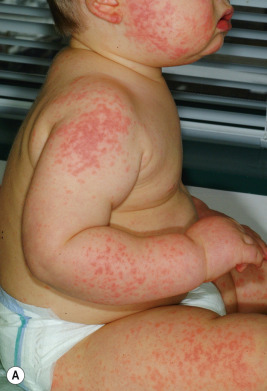
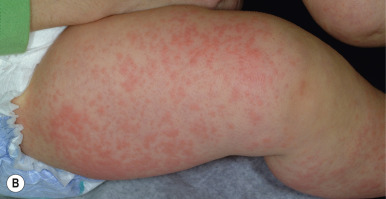
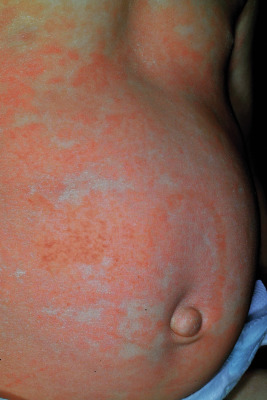
Most commonly, an exanthematous eruption begins 7 to 14 days after initiation of a new medication and sometimes after drug discontinuation. If an individual is rechallenged after an initial course of taking a medication, the reaction may develop within a few days. Most commonly, the eruption is symmetric and begins initially on the trunk before becoming generalized. Mucous membranes are usually spared, but palms and soles are often involved. Patients may experience varying degrees of associated pruritus and some have low-grade fever. Not uncommonly the eruption turns more of a brownish-red color in 7 to 14 days and may desquamate.
Penicillins, sulfonamides, cephalosporins, and antiepileptics are the most likely categories of drugs to cause exanthematous reactions. Viral exanthems tend to be indistinguishable from exanthematous drug eruptions and more commonly occur in pediatric patients. Eosinophilia favors a diagnosis of drug reaction; biopsy is generally not helpful. More severe drug reactions may be heralded by facial edema or marked eosinophilia, as in systemic hypersensitivity syndrome, or by mucous membrane lesions and dusky skin, as in SJS or toxic epidermal necrosis.
Treatment is largely supportive. The decision to discontinue a drug must be made based on the need for continuing the medication. Many patients will show clearance of the eruption despite continuation of medication (for example, when an infant with otitis is treated with amoxicillin). However, patients may show progression to erythroderma. Desensitization has been used for reactions to sulfonamides in patients with HIV infection.
Drug Reaction with Eosinophilia and Systemic Symptoms
This reaction, which is also called drug hypersensitivity syndrome ( DHS ), presents as an exanthematous drug reaction in association with fever, facial swelling, conjunctivitis, and often internal organ involvement that leads to mortality in 10% of patients. The clinical manifestations of DRESS syndrome typically begin 1 to 6 weeks after initiation of a responsible medication.
Fever and malaise often appear first and may be seen in association with cervical lymphadenopathy and pharyngitis. The cutaneous eruption occurs in approximately 75% of patients. It often starts on the face with edema, especially periorbital, then erythema and pruritus ( Fig. 20-19 ). The erythema then spreads caudally. The exanthematous eruption raises concern about SJS or TEN; the lack of mucosal involvement in DRESS can be a useful distinguishing feature.
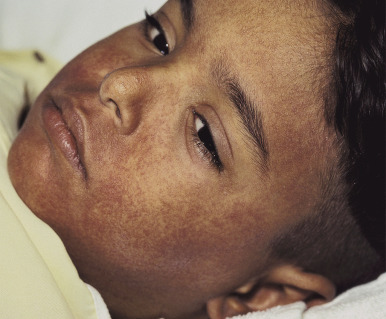
Of visceral sites of involvement, the liver is most common (about 50%), and the hepatitis may be fulminant, leading to death. Lymphadenopathy is often seen, and patients may complain of joint pain. Inflammation of the kidney, central nervous system, heart, and lungs has often been described. Thyroiditis occurs in a subset of patients and may not be noted until 2 to 3 months after onset. Atypical lymphocytosis and eosinophilia are seen in most affected individuals early in the course.
DRESS is most commonly caused by trimethoprim-sulfamethoxazole and the aromatic anticonvulsant agents, although viral infection has also been linked to DRESS. Sulfonamides are metabolized by acetylation to a nontoxic metabolite for renal excretion; slow acetylation has been associated with an increased risk of developing DRESS. Only aromatic amine forms of sulfonamides require this mechanism (e.g., these reactions do not occur with sulfonylureas and furosemide). Aromatic antiepileptics are metabolized by the cytochrome P450 system, and hereditary alterations in metabolism may lead to the accumulation of toxic arene oxide metabolites. Because these medications cross-react, affected individuals should avoid phenobarbital, phenytoin, carbamazepine, oxcarbazepine and lamotrigine. Alternative antiepileptics include valproic acid, benzodiazepines, gabapentin, zonisamide, topiramate, levetiracetam, and vigabatrin. Other drugs that less commonly cause this syndrome are minocycline, amoxicillin, dapsone, vancomycin, abacavir, nevirapine, aspirin, allopurinol, azithromycin, and traditional Chinese medicine. Reactions to doxycycline have not been described in patients who develop DRESS in response to minocycline. Clinical signs may vary dependent on causative drug. For example, lamotrigine rarely causes eosinophilia, abacavir leads to primarily respiratory and gastrointestinal manifestations without eosinophilia or hepatitis, and allopurinol reactions often involve the kidney.
The eruption of DRESS syndrome persists for weeks to months after medication withdrawal. After an initial period of improvement, the cutaneous and visceral manifestations of DRESS may flare 3 to 4 weeks after onset. Lymphocyte transformation tests may be useful in confirming the triggering medication but should be performed 5 to 8 weeks after onset in patients with DRESS. Challenge with the offending drug after initial reaction leads to reactivation of the fever and erythroderma within hours after initiation.
A role for concurrent herpes family viral infections has been emphasized in adult DRESS, particularly with reactivation of EBV and human herpesvirus (HHV) 6. A recent pediatric study found concomitant evidence of HHV6 infection in 50% of the children, and in association with greater pulmonary involvement (50%) and longer duration of fever and hospital stay.
Patients with suspected DRESS should have a variety of laboratory tests to consider visceral involvement. These include complete blood count, hepatic transaminases, serum creatinine level, urinalysis, and thyroid testing (which should be repeated after 2 to 3 months). Although topical corticosteroids and antihistamines may quell the pruritus in mild cases, patients with visceral involvement (especially for hepatitis, myocarditis, and interstitial pneumonitis) should be treated with systemic corticosteroids (1 to 2 mg/kg per day) for a few weeks, with gradual taper thereafter. Systemic corticosteroid treatment tends to shorten the hospital stay and reduce febrile days. Anecdotal cases of successful treatment of DRESS with IVIG have been described. Counseling of family members is important, because first-degree relatives of individuals with DRESS have a higher risk of developing these drug reactions.
Stevens–Johnson Syndrome and Toxic Epidermal Necrolysis
Stevens–Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) are now considered to be variants of the same hypersensitivity disorder. The incidence of these conditions is 1 to 3 per million persons per year, although in patients with HIV infection the incidence is 1 per 1000 persons per year. In a study of 708 patients, approximately 18% were children. Classification has been based on the degree of epidermal detachment. Epidermal detachment of less than 10% of the total body surface area is considered SJS; more than 30%, TEN; and between 10% and 30%, a transitional SJS-TEN overlap condition. In children, SJS is seen more often than TEN or SJS-TEN overlap; in one study, 85% of pediatric cases had SJS, whereas 6% had SJS-TEN overlap and 9% had TEN. Patients may appear to have SJS initially, but it evolves into typical TEN ( Table 20-3 ). At one time, SJS was lumped with EM (see Erythema Multiforme section), which is now considered to be a distinct entity, primarily resulting from hypersensitivity to herpes simplex infection. Similarly, TEN was once confused with staphylococcal scalded skin syndrome (SSSS), a more superficial and crusted disorder in which the superficial blistering is causes by an exfoliatin produced by Staphylococcus aureus (see Chapter 14 ).
| SJS | SJS-TEN | TEN | |
|---|---|---|---|
| Lesional morphology | Targetoid lesions, dusky red macules, bullae | Targetoid lesions, dusky red macules, bullae | Targetoid lesions, dusky erythematous macules and plaques; detachment of epidermis |
| Localization of skin lesions | May be scattered and isolated; may be confluent, especially on the trunk and face | May be scattered and isolated; often confluent | Usually extensive involvement with widespread confluence |
| Involved skin | <10% | 10% to 30% | >30% |
| Biopsy features | More interface dermatitis | Significant interface dermatitis + necrolysis | Predominantly necrolysis |
| Mucosal changes | Prominent | Prominent | May be less than in SJS |
| Systemic involvement | Often present | Always present | Always present |
The underlying triggers of SJS and TEN are similar, although drugs cause the majority of cases overall and almost all cases of TEN ( Box 20-2 ). In children, SJS is much less commonly caused by medication than in an adult. Most patients show evidence of SJS or TEN 7 to 21 days after the first drug exposure. Occurrence is almost always within the first 8 weeks of drug use and rarely within the first few days of drug administration. Children with an earlier onset (mean 2 to 3 days) have been previously exposed to the drug or a cross-reacting analogue. Although more than 200 medications have been implicated as potential triggers of SJS-TEN, sulfonamides, penicillins, phenobarbital, carbamazepine, and lamotrigine have been most strongly associated with SJS-TEN. An algorithm for assessing drug causality in epidermal necrolysis (ALDEN) has been proposed and correlates well with case-control analysis results. Patients carrying HLA-B*15:02 , which is common in the Asian population, are at strongly increased risk for carbamazepine-induced SJS/TEN, and this population should have human leukocyte antigens (HLAs) screened before initiating antiepileptic drugs. Other HLA antigens are being recognized in non-Asian populations. Aromatic antiepileptics (phenobarbital, phenytoin, carbamazepine, lamotrigine) tend to cross-react and cannot be substituted for each other. Drugs with longer half-lives are associated with a higher incidence than those related but with shorter half-lives. NSAIDs, including ibuprofen, and acetaminophen may also be triggers and possibly exacerbants with other drugs, and are best avoided during hospitalization for SJS-TEN.
Allopurinol
Barbiturates
Carbamazepine
Lamotrigine
NSAIDs
Penicillins
Phenytoin
Sulfonamides
NSAID, Nonsteroidal anti-inflammatory drug.
Mycoplasma infection is the most likely infectious trigger of SJS in pediatric patients and may be associated with less severe disease than drug-induced SJS. Based on a recent literature review of more than 200 cases of mycoplasma-induced “SJS” and “EM” in children, an alternative diagnosis of “ Mycoplasma pneumoniae -induced rash and mucositis” has been suggested for this disorder, distinguished clinically by its prominent mucositis (oral, ocular, urogenital) and usually sparse cutaneous involvement. Other infections, neoplasia, autoimmune disorders, and vaccination have also been implicated, particularly herpes simplex virus, which is more typically linked to EM.
SJS and TEN are disorders of keratinocyte death (apoptosis), and granulysin is the main mediator. Micro-ribonucleic acid (miR)-18a-5p, which targets an apoptosis gene ( BCL2L10 ), has recently been identified as a serum biomarker that can distinguish early TEN from other drug eruptions Lymphocyte transformation tests have helped to identify the causative agents in some affected children, but in contrast to those for children with DRESS syndrome, need to be performed during the first week of the disorder. The risk of developing SJS and TEN is significantly increased in patients with HIV infection and who have a decreased capacity to detoxify reactive intermediate drug metabolites (e.g., slow acetylators or individuals with defects in epoxide hydrolase-mediated detoxification).
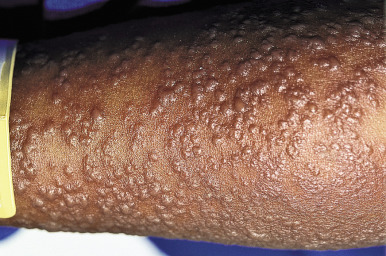
Older children and adults are most likely to develop this spectrum of disorders, but the condition has been described in young infants and neonates. High fever, pronounced constitutional symptoms, and varying degrees of generalized targetoid lesions, bullae, epidermal detachment, and mucosal erosions (of at least two sites) are characteristic (see Table 20-3 ). Patients not uncommonly show a 1- to 14-day prodromal period before the abrupt eruption of the typical features. During this prodromal period, affected children may show fever, malaise, headache, cough, coryza, sore throat, vomiting, diarrhea, chest pain, myalgia, and arthralgias.
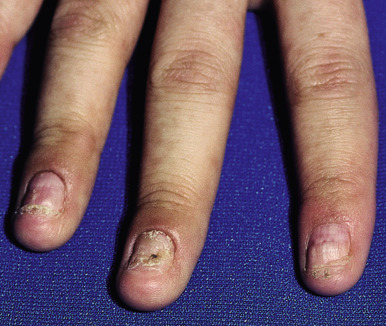
Cutaneous involvement most often appears initially on the face and upper trunk. Palms and soles are commonly involved ( Figs. 20-20 and 20-21 ). Erythematous and purpuric macules may develop flaccid gray discoloration or bullae ( Figs. 20-22 and 20-23 ), become confluent, and sometimes detach, leaving a raw, denuded base ( Figs. 20-24 and 20-25 ). Some of the macular lesions may show a dusky center, which gives a target-like (targetoid) appearance; however, the characteristic concentric rings of EM are absent. Some patients show limited targetoid lesions and little detachment (more typical of SJS), and others show extensive detachment (more typical of TEN). The early morbilliform eruption of TEN may be localized but more often is an extensive, painful erythroderma. After exerting light mechanical pressure with a finger to an area of erythema, the epidermis in patients with either SJS or TEN becomes wrinkled and peels off like wet tissue paper (see Fig. 20-25 ), the characteristic Nikolsky sign. The Nikolsky sign may be seen in patients without clinical evidence of epidermal detachment and can be a useful diagnostic tool. In addition to SJS and TEN, however, the Nikolsky sign may be seen in a variety of other bullous disorders (especially pemphigus, epidermolysis bullosa [see Chapter 13 ] and the SSSS [see Chapter 14 ]). Histopathologic examination of affected skin demonstrates necrosis of the lower epidermal cells with a sparse mononuclear cell infiltrate, and in the case of TEN, extensive necrosis with a subepidermal split. The epidermal necrosis correlates with the dusky blue coloration of lesions. The nails become dystrophic because of nailbed inflammation ( Fig. 20-26 ).
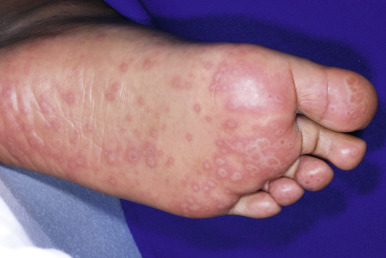
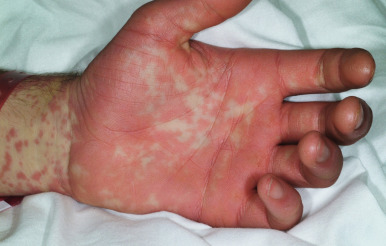
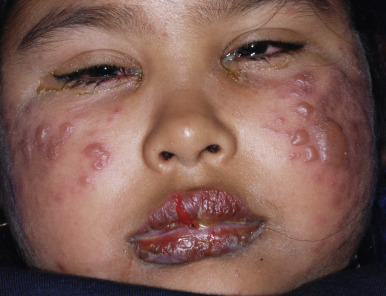
Extensive mucosal involvement is more typical of SJS than TEN. The mucosal manifestations of SJS tend to occur 1 to 2 days before cutaneous manifestations. The mucous membranes of the lips, tongue, buccal mucosae, eyes, nose, genitalia, and rectum may show extensive bullae with grayish white membranes, characteristic hemorrhagic crusts, and painful superficial erosions and ulcerations ( Figs. 20-27 and 20-28 ). Uncommonly, the esophageal and respiratory epithelial mucosae are affected. By definition, two or more mucosal surfaces are involved. The oral mucosa is always affected, resulting in inability to drink or eat and leading to a risk of dehydration. Genital area lesions lead to painful micturition and defecation. Although lesions of the oral mucosae tend to heal without scarring, strictures or stenosis of esophageal, vaginal, urethral, and anal mucosae have been described as sequelae.
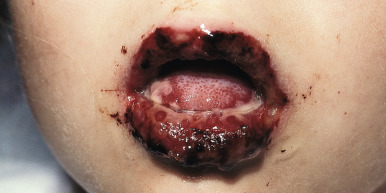
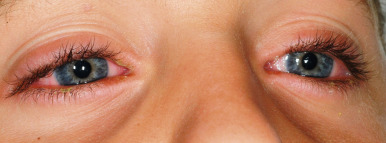
Severe purulent conjunctivitis with photophobia is the typical ocular manifestation (see Fig. 20-28 ). Corneal ulcerations, keratitis, uveitis, and panophthalmitis may also occur. Sequelae occur in 40% of patients and may be grave, with a possibility of keratoconjunctivitis sicca, corneal ulceration or neovascularization, trichiasis, symblepharon, and partial or even complete blindness. The severity of the acute ophthalmologic manifestations does not correlate with long-term complications. Pulmonary involvement may occur as an extension from the oropharynx and tracheobronchial tree or may be the result of pneumonitis associated with an initiating viral infection or secondary infection. In extreme cases, renal involvement with hematuria, nephritis, and in some cases, progressive renal failure may result. Esophageal or tracheal ulceration, pyoderma, lymphadenopathy, hepatosplenomegaly with elevated transaminase levels, myocarditis, arthritis and arthralgias, and/or septicemia may also complicate the disorder ( Box 20-3 ).
Constitutional
Fever
Dehydration
Mucocutaneous
Stomatitis with hemorrhagic crusts
Oral and genital erosions
Dysphagia
Purulent conjunctivitis with photophobia
Occasionally esophageal and pulmonary mucosal sloughing
Dusky erythematous macules, targetoid lesions, bullae, and skin sloughing
Visceral
Lymphadenopathy
Hepatosplenomegaly with hepatitis
Uncommonly: pneumonitis, arthritis, myocarditis, and nephritis
Laboratory abnormalities
Increased erythrocyte sedimentation rate (100%)
Leukocytosis (60%)
Eosinophilia (20%)
Anemia (15%)
Elevated hepatic transaminase levels (15%)
Leukopenia (10%)
Proteinuria, microscopic hematuria (5%)
SJS is most often confused with Kawasaki disease. However, the conjunctivitis of Kawasaki disease lacks exudation, and the mucosal erythema and dryness is not associated with hemorrhagic crust and mucosal denudation (see Chapter 21 ). The targetoid cutaneous lesions of SJS may resemble the target lesions of EM (see Erythema Multiforme section), a disorder usually attributed to herpes simplex infection in children; EM may also show blistering of the lip and oral mucosae that resembles SJS, although the blisters of EM are usually fewer and less symptomatic. The cutaneous lesions of EM are more often found on the extremities and/or face, whereas the atypical target or purpuric lesions of SJS are more commonly located on the trunk. Children with the rare immunobullous disorder, paraneoplastic pemphigus, may exhibit the mucocutaneous manifestations of SJS or TEN; the presence of epidermal acantholysis in biopsy specimens and the demonstration of antibody deposition in indirect immunofluorescence testing or immunoblot evaluation of serum allows paraneoplastic pemphigus to be distinguished (see Chapter 13 ). Other immunobullous disorders may also be confused with TEN (see Chapter 13 ). TEN and SSSS are usually easily distinguished; the blisters of TEN lead to denudement with keratinocyte apoptosis, whereas the desquamation of SSSS is superficial. In SSSS, the staphylococcal exfoliatin targets desmoglein 1, a desmosome component; because desmoglein 3 in the lower epidermis is able to compensate for desmoglein 1, the blistering of SSSS is superficial rather than panepidermal. SSSS also shows a different distribution with periorificial predominance and not mucosal involvement. Features of TEN occur as a severe manifestation of acute graft-versus-host disease, which is otherwise distinguished by history, other clinical findings, and histologic findings (see Chapter 25 ).
Although mild cases of SJS may show significant improvement within 5 to 15 days, the course of SJS or TEN is often protracted and may last more than a month. Epidermal detachment may be extensive, leading to massive fluid loss similar to that of a patient with burns. Bacterial superinfection with sepsis, impairment of temperature regulation, severe dehydration, electrolyte imbalance, excessive energy expenditure, and alteration in immunologic function are the usual complications. End-organ failure can be severe despite adequate supportive therapy. In adults, the mortality rate of SJS is 5%, transitional SJS-TEN 10% to 15%, and TEN 30% to 35%. Rates in children have been lower. A composite score (SCORTEN) has been used in adults to predict mortality and has recently been shown to be predictive in pediatric patients of mortality, length of hospital stay, and infectious complications, especially when scored on admission.
SJS and TEN are life-threatening disorders, and patients should be hospitalized. If damage to the epidermis is extensive, patients should be admitted to an intensive care unit or burn unit to allow special attention to fluid requirements, electrolyte balance, intravenous caloric replacement, and avoidance of secondary infection. If possible, all drugs administered within 2 months before onset of the eruption should be discontinued, and infectious causes should be sought and treated. Severe oropharyngeal involvement often necessitates frequent mouthwashes and local application of a topical anesthetic. When ocular involvement is present, ophthalmologic consultation should be obtained. Most recently, short-term use of potent topical corticosteroids and application of amniotic membrane to the entire ocular surface has been advocated to preserve visual acuity and an intact ocular surface. Patients should receive intravenous fluids and either a liquid diet, if tolerated, or parenteral nutrition; the measured resting energy requirement in pediatric SJS/TEN is increased by 30%. A mouthwash containing Maalox, elixir of diphenhydramine, and viscous lidocaine (mixed 1 : 1 : 1) can be soothing and decrease pain. Oral histamines such as hydroxyzine or diphenhydramine can be beneficial, and more vigorous pain control is often necessary. Of note, TEN has also been associated rarely with acetaminophen ingestion in children.
Manipulation should be avoided, but if necessary, should be performed in a sterile fashion. Intact areas of skin should be kept dry. Open wounds should be cleansed daily. Detached areas should be covered with nonadherent, moist dressings (such as petrolatum-impregnated gauze) until reepithelialization occurs. Periorificial areas can be treated with antibiotic ointment (such as bacitracin or mupirocin). Biologic dressings or skin equivalents may be considered for patients with extensive denudation.
The response to supportive care alone has been excellent in the majority of children. The administration of systemic medications as treatment is controversial, and there is little evidence-based data to support its use, especially in pediatric patients. Short-term administration of intravenous systemic corticosteroids (4 mg/kg per day) or pulsed intravenous steroids is sometimes advocated if started within the first 2 to 3 days of a drug-induced reaction. However, several anecdotal reports and open-label, noncomparative trials have not demonstrated efficacy. Continuing use of systemic steroids may increase morbidity and mortality from secondary infection, prolonging wound healing, masking early signs of sepsis, and triggering gastrointestinal bleeding. IVIG has been promoted as treatment of SJS and particularly TEN. Although some children with SJS or TEN have had shorter duration of fever and decreased development of new blisters with early administration (within 24 to 48 hours) of 2.5 to 3 g/kg per day IVIG for 3 days, other studies have shown no benefit to the use of IVIG, including for pediatric disease. Double-blind, randomized, controlled trials have not been performed. Cyclosporine, cyclophosphamide, and plasmapheresis have also been used anecdotally with good results. Thalidomide administration has been linked to increased mortality in a randomized placebo-controlled trial.
Long-term sequelae occur in 45% of affected children, including cutaneous dyschromia in 42%, which may persist for years. Sun exposure should be avoided and sunscreens used liberally for at least a few months because of the potential for ultraviolet light-induced worsening of the residual dyspigmentation. Persistent nail dystrophy or anonychia is also commonly reported (see Fig. 20-26 ). Late ocular complications include dry eye syndrome (59%), subjunctival fibrous scarring (33%), corneal erosions (29%), and in fewer than 25%, trichiasis, symblepharon, and visual loss. Artificial tears and lubricants may be required for several years after diagnosis, if not lifelong, because ocular disease tends to progress after hospital discharge. A high rate of recurrence of SJS in children has recently been disclosed, with approximately 20% of children readmitted at least once for recurrent episodes. These episodes occurred between 2 months to 7 years after the index episode and were more often related to drug ingestion than mycoplasma infection.
Fixed Drug Eruption
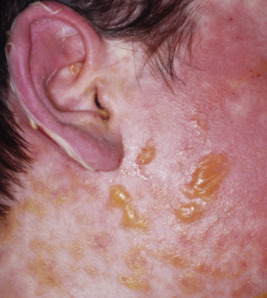
Fixed drug eruption is a term used to describe a sharply localized, circumscribed round or oval dermatitis that characteristically recurs in the same site or sites each time the offending drug is administered. The persistent residual hyperpigmentation leads to the name of the eruption, although occasionally lesions do not become pigmented. In one series, fixed drug eruption was second in incidence to exanthematous reactions to drugs and occurred in 22% of children with cutaneous reactions to drug ingestion. Lesions first occur 1 to 2 weeks after initial ingestion of the drug, but within 30 minutes to 8 hours after subsequent exposures. Fixed drug eruptions are type IV immune reactions and have recently been shown to result from CD8+ memory T cells that persist intraepidermally at site of involvement and, upon stimulation by drug, release interferon (IFN)-γ, leading to inflammation. Fas–Fas ligand interactions lead to the demonstrated basal keratinocyte apoptosis.
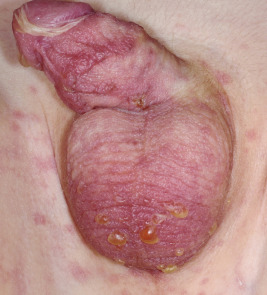
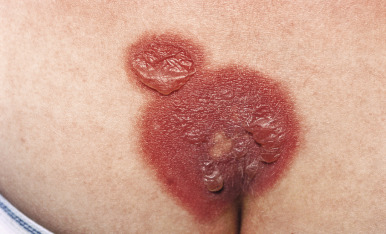
Lesions are solitary at first, but with repeated attacks, new lesions usually appear and existing lesions may tend to increase in size to more than 10 cm in diameter. Not uncommonly, lesions recur at the same site(s). The lesions tend to be erythematous, edematous, and dusky at their onset with well-defined borders. At times they may become bullous ( Fig. 20-29 ), with subsequent desquamation or crusting and a residual hyperpigmentation that may persist for months ( Fig. 20-30 ). Lesions can occur anywhere on the body but have a predilection for the perioral area, lips, hands, trunk, and genital region. In a series of affected boys with genital involvement, the clinical presentation usually consisted of swelling and erythema of the penis and/or scrotum associated with pruritus or burning, restlessness, urinary retention, and painful micturition.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


