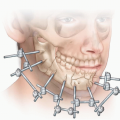
Spring-Assisted Surgery for Sagittal Synostosis
Lisa R. David
Claire Sanger Dillingham
DEFINITION
Craniosynostosis is the premature fusion of one or more cranial sutures (FIG 1).
Sagittal synostosis is the premature fusion of the sagittal suture, which leads to a long narrow-shaped head with frontal and occipital bossing due to the restriction in lateral growth as predicted by Virchow law.1
It is the most common form of craniosynostosis accounting for 55% of cases and is called scaphocephaly.2
ANATOMY
The skull is made up of bones that abut at sutures that allow growth of the underlying brain.
The primary sutures in an infant skull are the sagittal, coronal, metopic, lambdoid, and squamosal.
The sagittal suture is a dense, fibrous connective tissue joint that lies between the two parietal bones of the skull.
The bregma is the intersection between the sagittal and coronal sutures often referred to as the anterior fontanelle at birth.
The posterior portion of the sagittal suture ends at the intersection of the lambdoid sutures.
The point between these two is the vertex of the skull and is often the highest point on the skull.
PATHOGENESIS
Most craniosynostosis cases are not associated with a syndrome, although over 100 syndromes have been connected to this condition.3
Single-suture sagittal synostosis is the most prevalent type of craniosynostosis with an incidence of 1 in 5000 births.4
There is a higher observed incidence in boys compared to girls with a 4:1 ratio, respectively.5
Genetic mutations in the cell surface receptor fibroblast growth factor receptor (FGFR) 1 to 3 have been linked to the development of nonsyndromic craniosynostosis and syndromic cases such as Apert, Pfeiffer, Jackson-Weiss, Crouzon, and Muenke.6
Mutations identified in the targets TWIST1, bone morphogenetic proteins (BMPs), and RUNX2 have been linked to craniosynostosis.
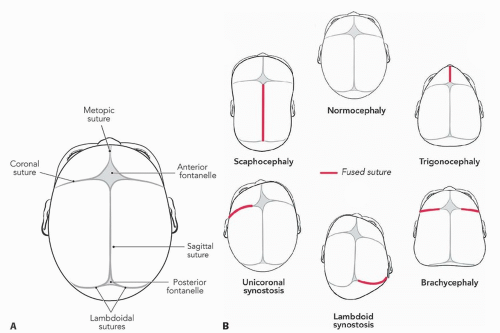
FIG 1 • A. Cranial sutures. B. Most common types of craniosynostosis and their associated shape change. (© WFBHS Plastic Surgery Collection.)
Transforming growth factor β (TGFβ) mutations have been involved in suture fusion but have not yet been linked to a syndrome.
Most craniosynostosis syndromes have an autosomal dominant inheritance.
NATURAL HISTORY
The time period of the most rapid brain growth in healthy humans is from the third trimester of pregnancy to the first year of life.
The average cranial capacity at birth is 350 cm4 and triples by 9 months of age.
In the absence of pathology, such as craniofacial malformations or malnutrition, the head circumference relates directly to the brain size, protein and DNA content, and the number of neurons.
Most cranial sutures remain open until several years of age with the exception being the metopic suture that can close as early as 1 year.
Most commonly, the sagittal suture begins to close from the posterior to anterior direction in the late second decade of life with complete closure seen at 35 years of age.
Sagittal synostosis can be detected as early as the 16th week of gestation by magnetic resonance imaging (MRI) and sonography.7
Sagittal synostosis is most commonly diagnosed in the first year of life.
Patients with sagittal synostosis are typically referred to the craniofacial surgeon after the pediatrician observes one or more of the following: the narrow and elongated head shape or frontal and occipital bossing, or the parents have expressed concern regarding the skull shape.
The incidence of elevated intracranial pressure (ICP) is low in patients with single-suture fusion; however, it is possible and should be considered.
Neurocognitive deficits are linked to craniosynostosis especially with multiple suture involvement.
Treatment aims to restore the calvarial shape, reduce ICP if needed, enhance development, and improve the appearance.
PATIENT HISTORY AND PHYSICAL FINDINGS
A thorough history and physical exam can provide information that leads to a diagnosis of associated conditions and avoids errors in failure to address significant concerns prior to surgery.
Perinatal history
Gestational age (this shape can be seen in prematurity without synostosis)
Age of parents
History of previous maternal miscarriages
Multiple births such as twins or triplets
Any perinatal distress
C-section vs natural vaginal delivery or assisted delivery with a device
Time spent in the ICU, intubated, or immobilized
Feeding history and if any intolerance is noted
Family history
Family members with any craniofacial anomalies or syndromes
Siblings of an affected child have double the estimated risk than the general population.
Physical examination
The cephalic circumference and index are measured and documented as they will be followed postoperatively.
Documentation should include intraocular distance, any signs of exorbitism, airway compromise, midface hypoplasia, or limb anomalies.
Evaluation for cleft lip or palate
An ophthalmology evaluation documenting the presence of increased ICP leading to optic nerve swelling, which results in papilledema
Consultation with a geneticist is warranted.
IMAGING
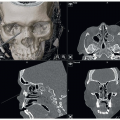
A computed tomographic (CT) scan provides valuable information as well as documentation of the condition (FIG 2).
It may also reveal additional pathologies that would have otherwise been hidden.
An MRI is not part of the standard imaging assessment in single-suture craniosynostosis but may be indicated if there is evidence of increased ICP.
The presence of agenesis of the corpus callosum or ventriculomegaly should prompt further evaluation.
A portable laser 3D scanning device is utilized at our institution and, if available, provides the team and family an objective analysis of cephalometric measurements, including cranial index (CI), 3D shape, and cranial volume calculated using Delta Scan Comparison software.
DIFFERENTIAL DIAGNOSIS
Single-suture craniosynostosis
Multiple suture craniosynostosis
Scaphocephalic head shape with no synostosis seen with significant prematurity
Positional plagiocephaly
Secondary craniosynostosis due to shunted hydrocephalus, lack of brain push, or trauma
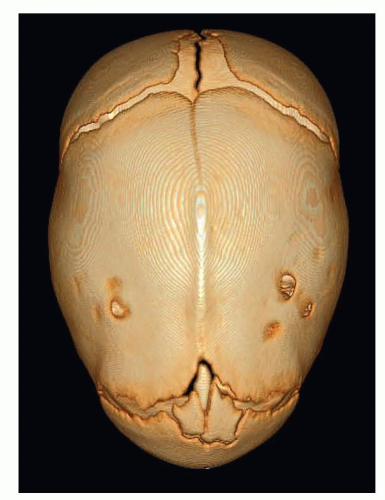 FIG 2 • 3D CT scan showing fusion of the sagittal suture. (© WFBHS Plastic Surgery Collection.) |
NONOPERATIVE MANAGEMENT
Nonoperative treatment of craniosynostosis is not recommended.
If there is a more serious life-threatening diagnosis, surgery for craniosynostosis should be delayed or undertaken with complete risks and benefits discussed with the family.
SURGICAL MANAGEMENT
Operative intervention for craniosynostosis is widely accepted and is the standard of care for these conditions.
The indication for surgical intervention for sagittal suture synostosis is full fusion of the suture or partial fusion with skull dysmorphology or increased ICP.
The ideal age for spring-assisted surgical correction is between 3 and 7 months of age.
This provides enough bone stock to handle the strength of the spring and a strong enough spring force to expand the skull bones.
This age range also allows for native bone regeneration and correction of further skull growth.
Risks to the patient include a scar on the scalp, bleeding, infection, spring dislodgement, cranial asymmetry, and need for further surgery.
Preoperative Planning
Preoperative evaluation should include a 3D CT scan of the skull to confirm the diagnosis of isolated sagittal craniosynostosis and rule out additional intracranial anomalies such as a Chiari malformation.
The CT also aids in operative planning for the selection of the spring strength and placement.
Positioning
The airway is secured, and ophthalmic ointment is placed in the eyes and secured with tape to keep the eyes closed and protected.
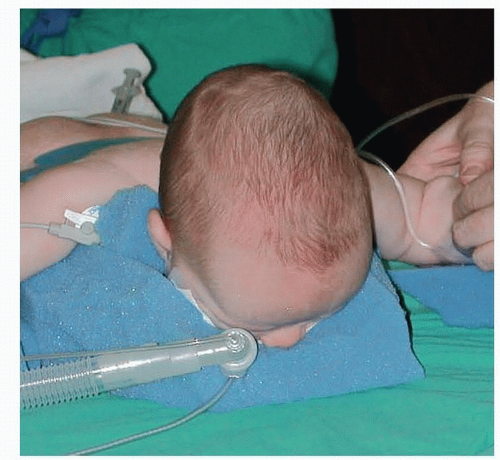
FIG 3 • Sphinx positioning with appropriate padding. (© WFBHS Plastic Surgery Collection.)Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree

 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access