Primary Immunodeficiency Disorders
Recurrent infections, including those involving the skin, raise the possibility that a child has an immune deficiency. The most common cause of immunodeficiency in children is acquired immunodeficiency resulting from human immunodeficiency virus (HIV) infection (see Chapter 15 ). Less commonly, children with evidence of an immunodeficiency have an inherited disorder. Genetic immunodeficiency disorders may show a variety of cutaneous abnormalities, some of which are unique and characteristic of the disorder, and others, such as dermatitis, that are shared by other immunodeficiencies and other disorders.
Some of these immunodeficiencies are discussed elsewhere because of their manifestations. Wiskott–Aldrich syndrome and hyperimmunoglobulinemia E (Hyper-Ig) syndrome are discussed in Chapter 3 , owing to the common presence of dermatitis. Chediak–Higashi and Griscelli syndromes, the silvery hair syndromes associated with immunodeficiency, are reviewed in Chapter 11 with the disorders of pigmentation. The telangiectasias that allow ataxia-telangiectasia to be distinguished from other forms of ataxia are described in Chapter 12 . Complement deficiencies are mentioned in Chapter 14 because individuals with a deficiency of a late complement component have an increased risk of neisserial infection, in Chapter 20 because hereditary angioedema (HAE) can be confused with angioedema, and in Chapter 22 because deficiency of the early components of complement may lead to a lupus-like disorder. Given its characteristic recurrent and recalcitrant candidal infections, chronic mucocutaneous candidiasis is reviewed in Chapter 17 .
Signs that should raise the suspicion of acquired or hereditary immunodeficiency are listed in Box 25-1 . Screening laboratory tests for a patient with recurrent cutaneous infections suspected of having an immunodeficiency are suggested in Table 25-1 .
History of infections
Increased frequency, severity, and duration
Unusual manifestations
Unusual infecting agents
Chronic infections, incomplete clearing
Poor response to appropriate agents
Severe viral infections
Recurrent osteomyelitis
Failure to thrive
Diarrhea, vomiting, malabsorption
Clues to specific types of immunodeficiency
Hematologic abnormalities (e.g., Wiskott–Aldrich syndrome)
Arthritis (e.g., Wiskott–Aldrich syndrome or early complement deficiency)
Paucity of lymph nodes (e.g., SCID) or lymphadenopathy (e.g., CGD)
Hepatosplenomegaly (e.g., CGD, Omenn syndrome, or Wiskott–Aldrich syndrome)
Poor wound healing (e.g., leukocyte adhesion deficiency)
Silvery hair (e.g., Chediak–Higashi or Griscelli syndromes)
CGD, Chronic granulomatous disease; SCID, severe combined immunodeficiency.
| Test | Primary Immunodeficiency |
|---|---|
| Complete blood count with differential, platelet count and examination of smear | Giant leukocyte granules (Chédiak–Higashi syndrome) |
| Thrombocytopenia (Wiskott–Aldrich syndrome) | |
| Leukocytosis (chronic granulomatous disease and leukocyte adhesion deficiency) | |
| Quantitative immunoglobulins | Selective IgA deficiency |
| Transient hypogammaglobulinemia | |
| X-linked hypogammaglobulinemia | |
| Hypogammaglobulinemia with hyper-IgM | |
| Common variable immunodeficiency | |
| Wiskott–Aldrich syndrome | |
| Hyperimmunoglobulin E | |
| Flow cytometry (T and B cells) | Severe combined immunodeficiency |
| Leukocyte adhesion defect (CD18 deficiency) | |
| Chronic granulomatous disease |
Immunoglobulin Deficiencies
Children with deficiencies of immunoglobulins (Igs) primarily manifest with bacterial infections beginning at 3 to 6 months of age, at a time when transplacentally derived maternal Igs wane. In general, the treatment of hypogammaglobulinemia is antibody replacement by intravenous infusions of immune serum globulin and vigorous antibiotic therapy.
The most common Ig deficiency is selective IgA deficiency, found in 1 in 500 individuals, of which 10% to 15% show clinical manifestations. About 5% of patients with IgA deficiency have mutations in TACI , which encodes a tumor necrosis factor (TNF) receptor family member. B cells in individuals with TACI mutations have impaired isotype switching and do not produce IgA and often IgG in response to TACI ligand. The most common features are sinopulmonary bacterial infections and Giardia gastroenteritis. Approximately one-third of patients with clinical manifestations develop immune-mediated disorders, some of which involve the skin. These include an atopic-like dermatitis, lupus erythematosus with selective IgA deficiency (recently linked to mutations in IFIH1 ), vitiligo, recurrent candidal infections, lipodystrophy, and idiopathic thrombocytopenic purpura. Two percent of individuals with celiac disease have full or partial selective IgA deficiency; this subgroup of patients has more concomitant autoimmune disorders and may have persistent elevation of IgG serologies, even with disease improvement. The risk of allergy is also increased with selective IgA deficiency, including asthma, cow’s milk allergy, and allergic rhinoconjunctivitis.
In addition to their decreased levels of IgA, 30% to 50% of affected individuals have serum anti-IgA IgG antibodies, leading to a risk of fatal anaphylactic reactions upon administration of blood products with IgA-bearing lymphocytes; if intravenous immunoglobulin (IVIG) is necessary, only IVIG with low IgA content should be administered.
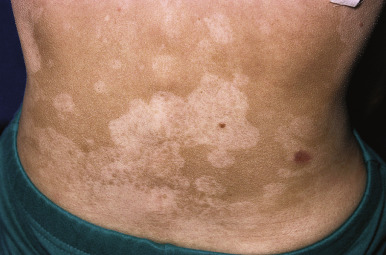
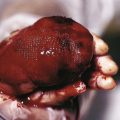
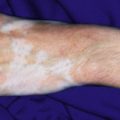
IgA deficiency may transition into (and is seen with increased incidence in families with) combined variable immunodeficiency (CVID), a heterogeneous group of disorders with decreased Ig levels (IgG, IgA, and sometimes IgM) and variable functional T-cell abnormalities. Patients with CVID most commonly show pyodermas, extensive warts, widespread dermatophyte infections, and dermatitis. They are predisposed to pyogenic infections of the upper and lower respiratory tract, as well as gastrointestinal infections, particularly those caused by Giardia . Noncaseating granulomas of the lungs, liver, spleen, and/or skin have been described and are not associated with microorganisms. Individuals with CVID also have an increased risk of autoimmune diseases, including vitiligo ( Fig. 25-1 ), alopecia areata, and vasculitis. The incidence of lymphoma is increased 400-fold, and that of cancer overall 8- to 13-fold. CVID is primarily a disorder of adults, with the mean age of onset 23 to 33 years ; however, 25% of cases are diagnosed before 21 years of age, with a peak incidence in children aged 5 to 10 years and a minimum age of 4 years used to exclude patients with other primary immunodeficiency disorders. Death in patients with CVID usually results from lymphoma, hepatitis, respiratory insufficiency, or gastrointestinal disease.
Most cases of panhypogammaglobulinemia in children represent X-linked hypogammaglobulinemia (also called X-linked agammaglobulinemia ), a disorder caused by mutations in BTK , which encodes a tyrosine kinase that regulates the conversion of pre-B cells to B cells able to differentiate and produce Igs. Less commonly, pediatric patients may have a transient form during infancy with early failure to thrive, protracted diarrhea, sinopulmonary infections, pyodermas, and cutaneous abscesses, but with reversal when Ig is produced at 18 to 30 months of age. Approximately 10% have an autosomal recessive form of panhypogammaglobulinemia.
Boys with X-linked hypogammaglobulinemia develop recurrent bacterial infections in the first year of life and have an increased susceptibility to hepatitis B and enteroviral infections. Furuncles and cellulitis are the most common infections. An atopic-like dermatitis is common, and noninfectious cutaneous granulomas have been described. There is an increased predisposition to the development of pyoderma gangrenosum, which has been recently linked to Warthin–Starry-positive Helicobacter bilis infection; although difficult to grow, the Helicobacter organisms are detectable by polymerase chain reaction (PCR) and electrospray ionization time-of-flight mass spectrometry. A small percentage of patients develop a dermatomyositis-like disorder with slowly progressive neurologic involvement, usually related to echoviral meningoencephalitis. Up to 6% of patients develop lymphomas.
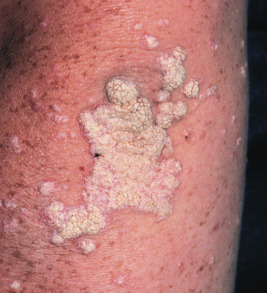
Patients with X-linked hypogammaglobulinemia with hyper-IgM (HIM) tend to have deficiencies of IgA, IgE, and IgG with neutropenia, but increased levels of IgM and isohemagglutinins. Rather than a primary B-cell defect, affected individuals have a primary T-cell defect. Cross-linking of CD40 on B cells induces switching of Ig classes from IgM to IgG, IgA, or IgE. Mutations in X-linked HIM lead to a dysfunction of the ligand for CD40 (CD40L). B cells from patients with HIM express functional CD40, but the T cells express the defective CD40 ligand and cannot bind CD40. Three other forms of HIM are autosomal recessive; these result from deficiency of CD40 itself, activation-induced cytidine deaminase (AICD), or uracil-N-glycosylase (UNG), the latter two being signaling components downstream of the CD40 receptor that are critical to B-cell differentiation and class switching. A rare form of X-linked immunodeficiency, sometimes associated with HIM, is associated with hypohidrotic ectodermal dysplasia (see Chapter 7 ), and has been linked to mutations in the NEMO gene, the same gene that is mutated in incontinentia pigmenti (see Chapter 11 ). Patients with HIM often seek treatment during infancy with bacterial and sometimes fungal or opportunistic sinopulmonary infections (most common feature), pyodermas, and gastrointestinal infections (especially with opportunistic infections such as Cryptosporidium ), hepatosplenomegaly, cervical adenitis, autoimmune disorders (especially thyroiditis, and hemolytic anemia), and an increased risk of lymphoma. Numerous, widespread warts ( Fig. 25-2 ), oral and perianal area ulcerations, and sclerosing cholangitis are additional features. Extensive warts are also a feature of warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM) syndrome, which results from mutations in the chemokine receptor CXCR4 ; EVER1 and EVER2 deficiency; DOC8 deficiency (see Hyperimmunoglobulinemia E section in Chapter 3 ); Netherton syndrome ( Chapter 5 ); GATA2 deficiency ; and STK4 deficiency.
X-linked lymphoproliferative disease (XLP) is characterized by an abnormal response to Epstein–Barr virus infection because of mutations in either SH2DIA , which encodes signaling lymphocytic activation molecule (SLAM)-associated protein (SAP) (XLP-1), or XIAP (XLP-2), critical proteins for cytotoxic T-cell function. Affected boys are healthy until they first develop infectious mononucleosis during childhood or adolescence. Fever, pharyngitis, maculopapular rash, lymphadenopathy hepatosplenomegaly, purpura, jaundice, and hemorrhagic colitis, and often hypogammaglobulinemia are typical features. The virus stimulates a rapidly progressive B-cell lymphoma, often with superimposed bacterial sepsis, which leads to death in 70% of affected boys, especially with hemophagocytic lymphohistiocytosis and without transplantation.
Chronic Granulomatous Disease
Chronic granulomatous disease (CGD) is a group of disorders characterized by severe recurrent infections resulting from an inability of phagocytic leukocytes to generate oxidative metabolites and activate neutrophil granule elastase and cathepsin G, thus blocking the killing of intracellular bacteria and fungi. In all forms of CGD the function of the nicotinamide dinucleotide phosphate (NADPH) oxidase complex is reduced. The disorder usually presents with recurrent pneumonias, hepatosplenomegaly, and lymphadenopathy. Patients develop granulomas, primarily of the lungs, liver, skin, and genitourinary and gastrointestinal tracts, as an abnormal immune response.
The most common form of CGD is X-linked recessive and results from mutations in the membrane-bound component of NADPH oxidase gp91 phox ( phox is short for phagocyte oxidase ), and 90% of affected individuals are male. Autosomal recessive forms (30% of patients) are due to mutations in other components of phagocyte NADPH oxidase, p22 phox and the cytoplasmic components p47 phox and p67 phox .
The skin, lungs, and perianal area are most often the sites of infection. Early lesions are usually cutaneous staphylococcal pyodermas and abscesses of the face and perianal area, not uncommonly associated with purulent dermatitis and regional lymphadenopathy. Seborrheic dermatitis, Sweet syndrome, scalp folliculitis, perioral and intraoral ulcerations that resemble aphthous stomatitis. Female carriers of gp91 phox mutations may show aphthous stomatitis and lupus-like eruptions, but rarely these have been described in affected patients with recessive form or administered Voriconazole.
The organisms associated with CGD are usually Staphylococcus aureus and opportunistic Gram-negative bacteria, including Serratia , Klebsiella , Pseudomonas , and Escherichia coli . These organisms all require oxidative metabolism for intracellular killing. Other organisms that may cause infection in patients with CGD with increased incidence are Aspergillus , Candida , Cryptococcus , Fusarium , and Nocardia . Bronchopneumonia and suppurative lymphadenitis are the most prevalent noncutaneous infections, and respond to appropriate antibacterial therapy and, in some cases, surgical drainage. The extracutaneous organs most commonly involved in CGD are the lymph nodes, lungs, liver, spleen, and gastrointestinal tract ( Box 25-2 ). CGD has been misdiagnosed as Crohn disease because of the overlap in features (failure to thrive, diarrhea, colitis, bowel obstruction, perianal ulcerations and fistulas, anemia, and hypoalbuminemia), and as hyper-IgE syndrome because of associated elevation of IgE levels in a case with selective IgA deficiency.
Seen in more than 50% of patients
Lymphadenopathy, lymphadenitis
Hepatosplenomegaly
Bronchopneumonia
Failure to thrive
Seen in 25% of patients or less
Persistent diarrhea
Pleuritis or empyema
Septicemia or meningitis
Hepatic or perihepatic abscess
Osteomyelitis
Seen in fewer than 25% of patients
Perianal abscess
Periorificial dermatitis
Aphthous stomatitis
Conjunctivitis
Lung abscess
Peritonitis
Obstructive granulomas
Patients with CGD commonly show leukocytosis, anemia, elevated erythrocyte sedimentation rate (ESR), and hypergammaglobulinemia. Skin tests for delayed hypersensitivity, phagocytosis, and chemotaxis are normal. Carrier and affected patients are often detected by quantitative dihydrorhodamine flow cytometry, and the nitroblue tetrazolium (NBT) screening assay is now rarely performed (in which the oxidized yellow form of NBT is reduced to a blue formazan precipitate). Ferricytochrome C reduction assay is a quantitative assay that shows absence of the respiratory burst. Immunoblots can detect the selective loss of membrane phagocyte oxidase components. However, mutations that lead to deficiency of gp91 phox or p22 phox cannot be differentiated by immunoblotting, because both are components of cytochrome b 558 , and the entire cytochrome is absent if one is deficient; gene sequencing is required to confirm the mutation in gp91 phox or p22 phox .
Antibiotic treatment of infection with surgical intervention as needed, and prophylactic trimethoprim-sulfamethoxazole and itraconazole therapy have been used in most affected individuals. Short courses of systemic corticosteroids have been helpful for patients with obstructive visceral granulomas. Stem-cell therapy has led to reversal of the immunodeficiency in severe cases with fewer infections and better linear growth, but management of CGD without transplantation similarly prolongs survival during childhood.
Leukocyte Adhesion Deficiencies
The leukocyte adhesion deficiencies (LADs) are a group of three autosomal recessive disorders that affect the ability of neutrophils, cytolytic T lymphocytes, and monocytes to be mobilized into extravascular sites of inflammation. In most affected individuals (LAD1), mutations in CD18 occur, leading to deficiency or dysfunction of the β 2 subunit of integrins. The principal ligand for these integrins is ICAM1, which participates actively in neutrophil and monocyte chemotaxis and phagocytosis. More than 75% of patients with severe disease die by 5 years of age; more than half of the patients with moderate deficiency die between the ages of 10 and 30 years.
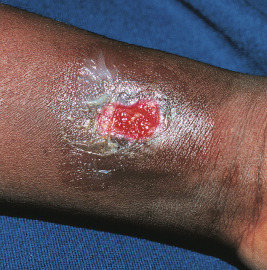
Patients with LAD have frequent skin infections (especially facial cellulitis and perianal infection), mucositis, and otitis, with a 5- to 20-fold increase in peripheral blood leukocytes. Poor wound healing is characteristic and leads to paper-thin or dysplastic cutaneous scars. Minor skin wounds may become large ulcerations that resemble pyoderma gangrenosum ( Fig. 25-3 ), especially if secondarily infected. Periodontitis is a typical feature, and if severe, may lead to loss of teeth. Bacterial and fungal infections may be life-threatening. Delayed separation of the umbilical cord is a common historic clue to the diagnosis. Psoriasis has been described in affected individuals with mild disease.
A second form of LAD (LAD2) is an autosomal recessive disorder caused by mutations in FUCT1. FUCT1 encodes a GFP-fucose transporter that is required for formation of sialyl-Lewis X, a ligand for selectins on the surface of neutrophils. In addition to their elevated leukocyte counts and recurrent bacterial infections, patients have short stature, a distinctive facies, and developmental delay. A third type of LAD, which features a bleeding tendency, is caused by a mutation in FERMT3, which encodes kindlin-3 and prevents activation of β1, β2, and β3 integrins. Dermal hematopoiesis, leading to the “blueberry muffin” presentation, has been described in affected infants and children.
Therapy of the soft-tissue infections includes antimicrobial agents and, as appropriate, debridement of wounds. Scrupulous dental hygiene reduces the severity of the periodontitis. Death usually occurs by 2 years of age in patients with severe LAD unless successful bone marrow transplantation or cord blood transplantation is performed. Oral fucose has been helpful for some patients with LAD2.
Severe Combined Immunodeficiency
Severe combined immunodeficiency (SCID) is a group of disorders with similar clinical manifestations and immune dysfunction, but different biochemical, cellular, and molecular features ( Table 25-2 ). Overall, 75% of affected patients are boys. The majority of cases are autosomal recessive. Approximately 46% have the X-linked recessive form resulting from mutations in gene that encodes γ c , which leads to an absence of T and natural killer (NK) cells but normal B-cell numbers. This γ c chain is a component of several interleukin receptors and is critical for T- and NK-cell function. Mutations in adenosine deaminase (ADA) lead to the second most common form of SCID. Accumulation of adenosine when its deaminase is missing is toxic to lymphocytes. Omenn syndrome usually results from deficiency of recombination activating gene (RAG) proteins, which mediate the deoxyribonucleic acid (DNA) double strand breaks that allow variable (diversity) joining (V[D]J) recombination and Ig diversity.
| Disorder | Gene Location | Gene | Diagnostic Tests | Cells |
|---|---|---|---|---|
| ADA deficiency * | 20q12-13 | ADA | Red cell ADA levels and metabolites | T − /B − /NK − |
| Artemis deficiency | 10p13 | Artemis | Defects in V(D)J recombination; increased sensitivity to radiation | TB − /NK + |
| CD45 deficiency | 1q31-32 | CD45 | CD45 expression | T − /B + /NK |
| IL-7 receptor deficiency | 5p13 | IL-7 receptor α | IL-7 receptor α expression | T − /B + /NK + |
| JAK3 deficiency | 19p13 | JAK3 | JAK3 expression/signaling | T − L/B + /NK − |
| MHC class II deficiency | 16p13 | CIITA | HLA-DR expression | T + /B + /NK + |
| 1q21 | RFX5 | |||
| 13q13 | RFXAP | |||
| PNP deficiency | 14q11 | PNP | Red cell PNP levels and metabolites | T − /B − /NK − |
| RAG deficiency, Omenn syndrome | 11p13 | RAG1 and RAG2 | Defects in V(D)J recombination; T- and B-cell clonal analysis | T − /B − /NK + |
| T-cell receptor deficiency | 11q23 | CD3g | CD3 expression | T − /B + /NK + |
| X-linked SCID † | Xq13 | Common γ chain | γ c expression by FACS | T − /B + /NK − |
| ZAP70 deficiency | 2q12 | ZAP-70 | ZAP-70 expression | T − /B + /NK − |
* Most common autosomal recessive form.
Infants may present have a generalized seborrheic-like dermatosis, a morbilliform eruption, or exfoliative erythroderma with alopecia. Extensive cutaneous inflammation is a characteristic of 98% of infants with Omenn syndrome, a subset of SCID with reticuloendothelial cell proliferation. Patients with Omenn syndrome typically show hepatosplenomegaly (88%), lymphadenopathy (80%), alopecia (57%), eosinophilia, and a high serum IgE level. Oral and genital ulcers are characteristic of defects in patients with an Artemis mutation, especially in Athabaskan-speaking American Indian children. Multicentric dermatofibrosarcoma protuberans and pulmonary alveolar proteinosis have recently been described in association with ADA deficiency.
Acute graft-versus-host disease (GVHD) from maternal cell engraftment or nonirradiated transfusions should also be considered in an infant with an extensive cutaneous eruption. Biopsy will allow differentiation. A more chronic form of GVHD (often with the acute form in utero ) may also present as lesions that resemble lichen planus, lamellar ichthyosis, or scleroderma.
Recurrent infections, diarrhea, and failure to thrive are apparent by 3 to 6 months of age. The most common early infections are candidal infections and pneumonia caused by bacteria, viruses, or Pneumocystis jiroveci . Patients with SCID usually lack tonsillar buds, thymus, and palpable lymphoid tissue, despite recurrent infections. Nearly all patients with SCID have a profound deficiency of T lymphocytes and a low absolute lymphocyte count. Patients are further classified by the results of fluorescent activated cell sorter (FACS) analysis into those with B lymphocytes (T − /B + SCID) and those without B lymphocytes (T − /B − SCID). Further subclassification can be made according to the presence or absence of NK cells (see Table 25-2 ). The specific diagnosis is confirmed primarily by direct gene analysis and flow cytometric analysis of peripheral blood mononuclear cells with antibodies directed against specific proteins missing from the cell surface, such as JAK3 or γ c. The natural outcome for SCID is poor, and most patients die by 2 years of age without intervention. Hematopoietic stem-cell transplantation in infants is the treatment of choice and leads to survival of 95% of infants if performed in the first 3 months of life. Bone marrow-derived CD34+ cells transduced with the SIN-γ c modified γ-retrovirus vector without preparative conditioning led to T-cell recovery without evidence of leukemia development after 12.1 to 38.7 months of follow-up study in nine boys with X-linked SCID. Early diagnosis of SCID is critical, preferably before the administration of live vaccines, nonirradiated blood products and, in countries where applicable, bacille Calmette–Guérin (BCG). Patients with SCID have a high risk of BCG-associated complications (34% disseminated and 17% localized) and subsequent death, especially if they are given the vaccination at 1 month of age or younger and with T-cell numbers of 250/mcL or less at diagnosis. In a United Kingdom-based study, diagnosis at birth because of a positive family history significantly improved outcome with survival of greater than 90% related to reduced rate of infection and improved transplantation outcome, suggesting that neonatal screening for SCID will also improve outcome. Perinatal screening is now mandatory in several states.
Graft-Versus-Host Disease
The host-versus-graft reaction occurs when a graft or transplant (skin, heart, or kidney) is placed in a normal individual and the host’s circulating immune cells react against the foreign tissue, causing graft or transplanted tissue destruction. In GVHD, the reverse happens. The inflammation from conditioning regimens is thought to activate host antigen-presenting cells and chemokines that recruit donor leukocytes into host target organs. Activated donor T cells stimulate dendritic cells, leading to further T-cell stimulation and expansion and culminating in target organ apoptosis and dysfunction. GVHD most commonly occurs in children with malignancy suppressed by radiation and chemotherapy, who receive hematopoietic stem-cell transplantation, or in immunodeficient children who receive nonirradiated blood products. The risk of both acute GVHD (aGVHD) and chronic GVHD (cGVHD) is greater in males who receive female donor cells because of responses to H-Y minor histocompatibility antigens. It has been estimated that moderate to severe GVHD occurs in 10% to 50% of patients given an allogeneic transplant from a human leukocyte antigen (HLA)-identical donor and much more commonly in patients given a transplant from a partially matched family donor or an unrelated volunteer. In utero GVHD may occur in immunodeficient babies exposed to maternal antigens. GVHD can occur in recipients of solid-organ transplants, especially multiorgan transplants, and reflects donor T-cell chimerism.
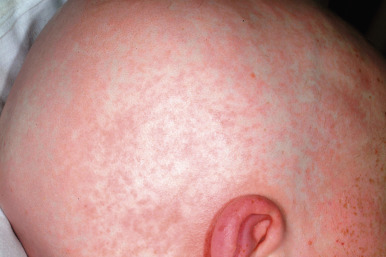
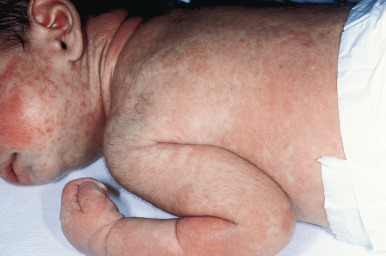
The response to the host can be early (acute) or late (chronic). aGVHD classically develops during the first 100 days (usually 2 to 4 weeks) but can be persistent, recurrent or late onset, and usually involves the skin, gastrointestinal tract, and liver (in that order of incidence). Patients who receive autologous transplants may have a mild cutaneous form of aGVHD that occurs 1 to 3 weeks after transplantation and resolves spontaneously. Reactions to transfusions often occur 7 to 10 days after the transfusion. The most common cutaneous manifestation of aGVHD is erythematous macules and maculopapules that often begin on the ears, face, neck, palms, and soles and then become generalized ( Figs. 25-4 and 25-5 ). The cutaneous eruption may become confluent and desquamate. Mild to moderate aGVHD most often presents with a nonspecific cutaneous eruption and thus can be confused with infection (especially viral), drug reaction, and reactions related to the transplant (e.g., the self-limited eruption of lymphocyte recovery, engraftment syndrome (which is characterized by associated fever and vascular leak with weight gain, edema, ascites, pulmonary infiltrates and hypotension), and toxic erythema of chemotherapy (which is painful and usually localized to the palms, soles and intertriginous areas. Severe aGVHD may resemble exfoliative erythroderma or toxic epidermal necrolysis. Patients with aGVHD may complain of nausea or crampy abdominal pain and have watery or bloody diarrhea, hepatomegaly, and hepatic function abnormalities. Many patients are anorexic and have fever.
Milder forms of aGVHD may be difficult to distinguish from an infection (especially viral) or drug reaction. Abnormalities seen in biopsy specimens reflect the clinical severity of the cutaneous manifestations and range from nonspecific changes to basal keratinocyte vacuolization with scattered necrotic keratinocytes surrounded by lymphocytes (the characteristic “satellite cell necrosis”) to severe epidermal necrosis. As such, biopsies are not helpful for patients with milder disease and do not tend to affect treatment decisions. Laboratory tests may show eosinophilia and elevation of bilirubin and hepatic transaminase levels.
cGVHD occurs in 6% (matched sibling cord blood) to 65% (matched unrelated donor peripheral blood stem-cell transplants) of pediatric patients. It typically presents 6 to 18 months after allogeneic stem-cell transplantation. Patients may have previous aGVHD that resolves or progresses directly into cGVHD, or may have no history of preceding aGVHD. cGVHD is now defined based on clinical and histopathologic signs of presentation, rather than time of onset since transplantation (formerly ≥100 days) and can be subclassified as mild, moderate, or severe based on number of sites, disability, and lung involvement. An overlap syndrome with features of both aGVHD and cGVHD has been described. cGVHD often resembles autoimmune disorders and in contrast to aGVHD, can affect virtually any organ, leading to significant morbidity with decreased quality of life and overall survival. Decreased survival of patients with cGVHD has been associated with thrombocytopenia, progressive onset, extensive cutaneous involvement, gastrointestinal involvement and a low Karnofsky performance status at diagnosis.
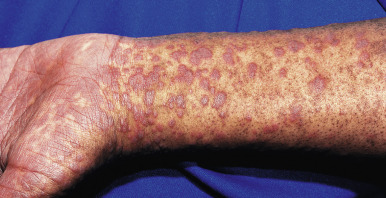
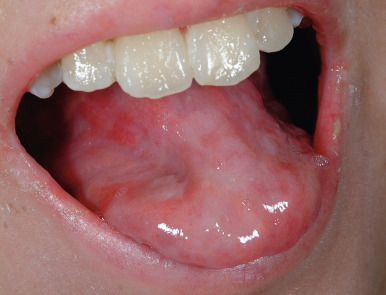
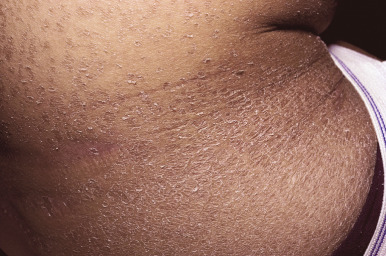
cGVHD often manifests with generalized cutaneous involvement ( Table 25-3 ). Early lesions may resemble lichen planus with flat-topped violaceous papules and plaques (including mucosal changes of lichen planus) ( Figs. 25-6 and 25-7 ) (see Chapter 4 ). Lichenoid lesions may be localized to Blaschko lines or to the site of previous herpes zoster infection. Some patients with cGVHD manifest acquired ichthyosis that resembles ichthyosis vulgaris ( Fig. 25-8 ) or lichen sclerosus ( Fig. 25-9 ). Later changes of cGVHD include generalized xerosis, patchy dyspigmentation ( Fig. 25-10 ), progressive poikiloderma, sclerodermatous changes with joint contracture and ulcerations ( Figs. 25-10 through 25-13 ), cicatricial alopecia ( Fig. 25-14 ), and nail dystrophy resembling that of dyskeratosis congenita (see Chapter 7 , Fig. 7-63 ). Disseminated porokeratosis has also been described after stem-cell transplantation and may respond to acitretin administration.
| Location | Characteristic Features | Other Features |
|---|---|---|
| Skin | Lichen planus-like Sclerotic (or lichen sclerosus-like) Poikilodermatous Ichthyotic Vaginal scarring, stenosis | Depigmentation Genital erosions, ulcers, fissures |
| Hair | Alopecia, nonscarring, or cicatricial | |
| Nails | Ridging, onycholysis, loss, pterygium | |
| Oral | Lichen planus-like Keratotic plaques Sclerosis with restricted opening | Mucosal atrophy Xerostomia Ulceration Mucocele |
| Eyes | Dry eyes Cicatricial conjunctivitis Keratoconjunctivitis sicca Punctate keratopathy | |
| Joints/muscle | Contracture from sclerosis Fasciitis, arthritis | Myositis |
| GI | Chronic diarrhea Upper esophageal strictures, stenosis, webbing Hepatomegaly | |
| Lungs | Bronchiolitis obliterans Interstitial fibrosis |
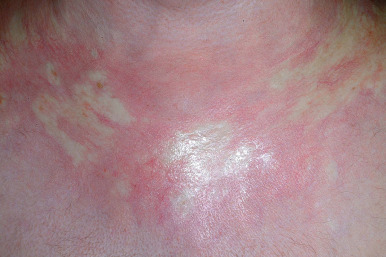
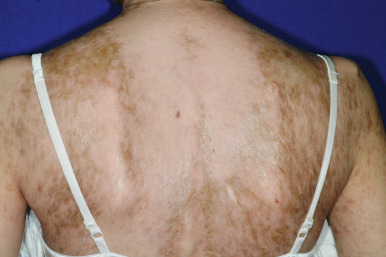
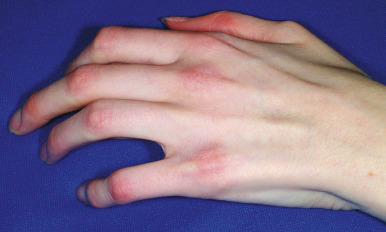
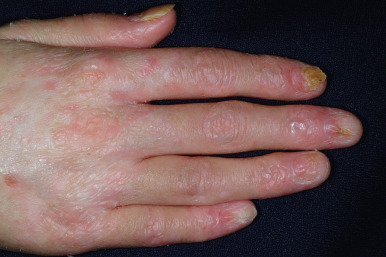
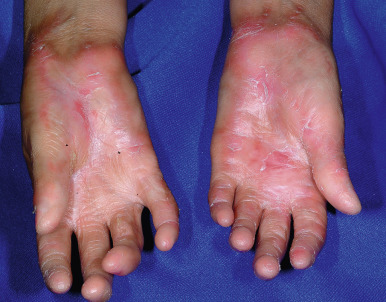
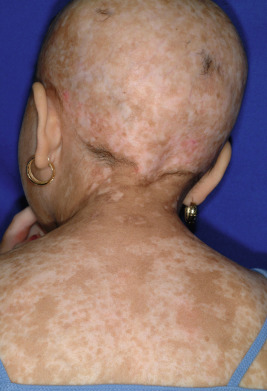
Affected individuals may have dry eyes (as in Sjögren syndrome; see Chapter 22 ) and desquamative esophagitis with stricture formation. Weight loss can result from dysphagia and mucositis. Other manifestations are chronic diarrhea, hepatomegaly, lymphadenopathy, myositis, arthritis, pleural and pericardial effusions, and pulmonary fibrosis.
Patients with cGVHD show evidence of immune dysfunction and have recurrent infections. The majority of patients with cGVHD have eosinophilia and hypergammaglobulinemia. Many have thrombocytopenia and increased titers of a wide variety of autoimmune antibodies, especially antinuclear antibodies (ANAs). Liver function testing may show evidence of cholestasis. Pulmonary function tests are abnormal in approximately 50% of patients, and chest radiographs may reveal interstitial fibrosis. Biopsies of skin lesions show changes consistent with the clinical picture. For example, patients with lichenoid lesions have the histologic changes of lichen planus (see Chapter 4 ), often with satellite cell necrosis. Patients with sclerosis tend to show band-like inflammation at the dermal–epidermal junction and dermal sclerosis with obliteration of adnexae. Direct immunofluorescence analysis of skin specimens often shows the linear deposition of immunoreactants at the dermal–epidermal junction.
The mortality rate of GVHD ranges from 12% for mild aGVHD to 55% for severe aGVHD, usually because of infection. The mortality of cGVHD is 25%, usually from infection, hepatic failure, and malnutrition. Having aGVHD is a major risk factor for development of cGVHD (11-fold increased risk), emphasizing the importance of preventing aGVHD. Other risk factors are an unrelated or mismatched donor, use of peripheral blood stem cells, older age of donor or recipient, use of total body irradiation, male recipient with a female donor, and malignant disease. For patients with myeloablation (full-intensity conditioning, which eliminates all malignant and marrow hematopoietic cells), prophylaxis usually involves 2 to 12 months of tacrolimus (or cyclosporine), with or without a short course (about 1 month) of methotrexate. With reduced intensity conditioning before transplant, which does not fully eradicate malignant and hematopoietic cells and instead relies on immunologic antihost and antitumor effects, a calcineurin inhibitor and mycophenolate mofetil (MMF) (or, instead, corticosteroid) are generally used prophylactically. In both situations, anti-T-cell serotherapy with either antithymocyte globulin or alemtuzumab (Campath) is usually added for unrelated donor transplants and also reduces graft rejection; for haploidentical donors, T-cell depletion or CD34 selection is often performed. Environmental isolation, bowel rest through hyperalimentation, irradiation of blood products, and prevention of infection are other protective measures.
If grade II through IV aGVHD develops, the first-line therapy is continuing prophylactic immunosuppression (optimizing calcineurin inhibitor levels) and adding methylprednisolone 1 to 2 mg/kg per day. For skin GVHD, topical steroid and/or tacrolimus may be added, with oral administration of budesonide for gastrointestinal GVHD. Patients who fail to respond to corticosteroid alone after 5 to 7 days can be treated with salvage therapy, such as MMF, anti-TNF antibodies (e.g., infliximab), mammalian target of rapamycin (mTOR) inhibitors (e.g., sirolimus/ rapamycin), anti-interleukin (IL) 2 antibodies (e.g., daclizumab), or extracorporeal photopheresis (ECP). Responses in general are poor if initial steroid treatment is not successful, and the risk of infection is high. If at least two second-line treatments fail, alternatives are alemtuzumab (Campath), methotrexate, pentostatin, and mesenchymal stem cells.
Similar regimens have been used to treat cGVHD, although there is no proven standard, with prednisone and calcineurin inhibitors most commonly used for first-line therapy. The mean duration of therapy for patients with cGVHD is 3 years, with about half of the patients able to discontinue therapy 5 years after transplantation. Second-line treatment (steroid-resistant or steroid-sparing) includes one of more of the following: ECP (which gives the best result for skin, oral or liver cGVHD), rituximab, imatinib mesylate (increases range of motion with sclerotic cGVHD), mTOR inhibitors, and pentostatin. Third-line treatment includes pulsed high-dose intravenous methylprednisolone, MMF, or methotrexate. Overall, the 5-year survival rate is 50% for those who respond to therapy for GVHD and 30% for those with no or an incomplete response. Mucocutaneous manifestations of GVHD may also benefit from administration of thalidomide, acitretin, application of topical tacrolimus, or treatments with narrowband ultraviolet B (UVB) light, psoralen plus ultraviolet A light, or UVA1 light. Supportive intervention for cGVHD includes supplemental nutrition (usually parenteral), scrupulous care of wounds, physical therapy to prevent joint contractures and disability, artificial tears, cutaneous emollients and sunscreens, and prophylaxis against Pneumocystis infection with trimethoprim-sulfamethoxazole.
Orofacial Granulomatosis
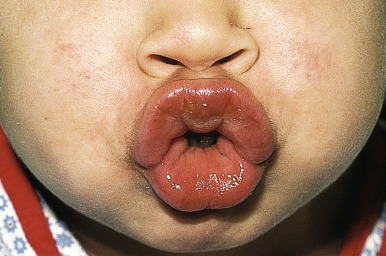
Orofacial granulomatosis (OFG; also called cheilitis granulomatosa or granulomatous cheilitis ) ( Fig. 25-15 ) is characterized by face and lip swelling (macrocheilitis). It is the most common manifestation (93% with facial swelling and 66% with lip swelling) of Melkersson–Rosenthal syndrome, which also includes recurrent facial paralysis and a furrowed or “scrotal” tongue (lingua plicata) ( Fig. 25-16 ). Furrowing of the tongue and facial palsy have each been reported to occur in approximately 30% of patients. No prodrome warns of attacks, and patients experience no associated erythema, pain, or pruritus. The disorder is more prevalent in males and has a mean age of onset of 11 years. The swelling can increase the size of the lip by twofold to threefold, leading to chapping from exposure of the mucosa. Both lips may be involved, and the swelling may be bilateral or unilateral. In addition to the swelling of the lips and other oral mucosal structures, the eyelids may be swollen. The attacks usually disappear within days or weeks but commonly tend to persist after several recurrences. Facial palsy usually occurs on the side of the facial swelling and tends to resolve spontaneously.
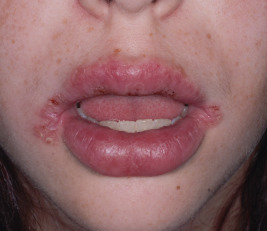
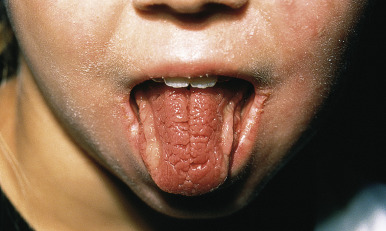
The cause is unknown. Attacks usually start during adolescence with paralysis of a facial nerve, repeated severe headaches, edema of the circumoral tissue of the upper lip or cheeks, and occasionally edema of the gingivae, sublingual area, and lower lip. The edema is usually asymmetric, but the whole face may be involved. Associated signs include hyperhidrosis, loss of taste, and visual impairment. Biopsy specimens show noncaseating granulomas in an edematous stroma that may be indistinguishable from the granulomas of Crohn disease, and the relationship between Crohn disease and OFG remains controversial. In one systematic review, gastrointestinal signs were present in 26% at time of OFG diagnosis, and 40% of children received a concomitant diagnosis of Crohn disease, with OFG preceding signs of Crohn disease in more than 50%. Concomitant perianal disease and a family history of Crohn disease are associated with a higher risk of Crohn disease.
Intralesional injections of triamcinolone or systemic administration of corticosteroids can control the facial and mucosal swelling; intermittent application of topical clobetasol gel may also lead to improvement. Minocycline, with or without corticosteroids, and sometimes macrolides have also been helpful. Other therapies include thalidomide, dapsone, azathioprine, clofazimine, and TNF-α inhibitors, particularly infliximab. Wedge resection of the inner lip should be reserved for patients who fail to respond to other options. The recurrent facial palsy tends to be more persistent than recurrent Bell palsy, and transmastoid subtotal facial nerve decompression can lead to improvement in most cases.
Crohn Disease
Crohn disease is a granulomatous disorder of the intestinal tract that usually has its onset between 15 and 30 years of age but has been described in young children. Approximately one-third of pediatric patients have small intestinal disease, one-third have ileocolitis, and one-third have colitis. Total colitis is more common than segmental or isolated proctitis. Growth failure, delayed pubertal development, and osteopenia are important complications of Crohn disease.
The pathogenesis of Crohn disease is complex. The disorder is thought to occur in genetically predisposed individuals when the intestinal mucosal immune function is altered by exogenous agents, such as infectious organisms, or host factors, such as intestinal barrier function, vascular supply, or stress. The disorder occurs twofold to fourfold more often in Ashkenazi Jews and has been linked to certain HLA alleles (DR3, DQ2, DR103), as well as to mutant alleles of NOD2/CARD15 . The CARD15 protein senses bacterial peptidoglycan and regulates nuclear factor (NF)-κB signaling. Granulomatous colitis is also a feature of a subset of individuals with Hermansky–Pudlak syndrome (see Chapter 11 ).
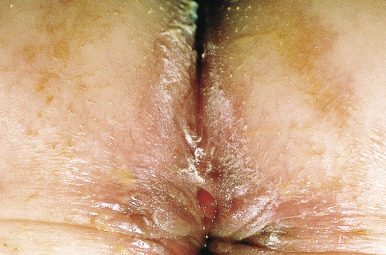
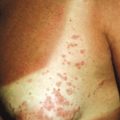
Skin manifestations may occur before, concomitantly, or after other evidence of disease. Perianal skin tags are found in 75% to 80% of patients with Crohn disease ( Fig. 25-17 ) and occur before gastrointestinal manifestations in 25% of patients. In addition to tags, skin and mucosal findings have been noted in 22% to 44% of patients ( Table 25-4 ) and occur more often in patients with colonic versus small intestinal disease. Several mucocutaneous manifestations of Crohn disease also are features of ulcerative colitis. Erythema nodosum (see Chapter 20 ) occurs in up to 15% of patients with Crohn disease and has been shown to correlate with the presence of arthritis. Pyoderma gangrenosum occurs less often in Crohn disease (approximately 1% of patients) ( Fig. 25-18 ) than in ulcerative colitis (up to 5% of pediatric patients), occasionally precedes the development of bowel symptoms, and has a course unrelated to the activity of the bowel disease. Oral aphthous ulcers have been described in greater than 50% of pediatric patients with Crohn disease. Pyostomatitis vegetans, erythematous pustular lesions associated with erosions and ulcerations, is considered a marker for inflammatory bowel disease. Cutaneous changes related to nutritional deficits (particularly acrodermatitis enteropathica caused by zinc deficiency) can occur as well.
| Specific | Nonspecific | |
|---|---|---|
| Cutaneous | Extraintestinal cutaneous Crohn disease | Erythema nodosum Pyoderma gangrenosum Polyarteritis nodosa Erythema multiforme Vasculitis |
| Anogenital | Perianal fissures and tags Sinus tracts Labial or scrotal edema | |
| Oral | Orofacial granulomatosis Cobblestoning Mucosal tags | Aphthous stomatitis Angular cheilitis |
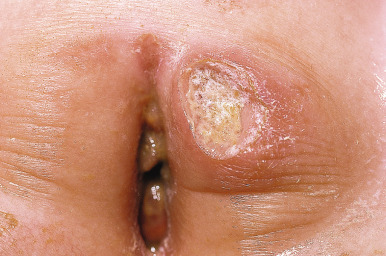
Other mucocutaneous manifestations are more specific for Crohn disease. Cobblestoning of the buccal mucosa is seen in up to 20% of affected individuals. Painless swelling of the lips, resembling OFG, may be the initial presentation of Crohn disease in a child or adolescent and may precede the gastrointestinal symptoms by several years ( Fig. 25-19 ). Similarly, the vulva may show erythema, swelling, and fissuring, and the scrotum and penis may be swollen and erythematous. Perianal lesions may extend onto the adjacent perineum, abdomen, or buttocks with fissures, and sinus tracts are common. Nasal perforation and chronic granulomatous otitis externa have been described. Abdominal surgical sites may also be loci for cutaneous involvement. Biopsy provides the clue to diagnosis, although the noncaseating granulomas resemble those of Melkersson–Rosenthal/cheilitis granulomatosa and sarcoidosis. Special stains (periodic acid–Schiff, Ziehl–Nielsen and Gram) are important to consider infectious causes of granulomas. These granulomatous lesions of the lips and anogenital area are considered “contiguous” Crohn disease because of their proximity to the gastrointestinal tract. Granulomatous lesions elsewhere are considered extraintestinal (sometimes called metastatic ) cutaneous Crohn disease. These lesions typically are dusky red, swollen/indurated plaques, most commonly found on the genital region of children, lower extremities, abdomen, or trunk; lesions less commonly show no erythema or are ulcerating.
Barium contrast studies are particularly important for investigating small bowel disease, but endoscopy and colonoscopy are important tools for upper and lower bowel disease, respectively. Initial treatment involves administration of systemic corticosteroids and aminosalicylates, with the early addition of azathioprine or 6-mercaptopurine as steroid-sparing alternatives for maintenance therapy. TNF-α inhibitors (etanercept, adalimumab, and infliximab) are very helpful as well, and reduce the need for surgery. The development of psoriasiform dermatitis is common in patients with Crohn disease treated with TNF-α inhibitors and often manifests in periorificial and genital areas, scalp, hands and feet (see Chapter 4 ; Fig. 4-25 ). The mechanism for the common occurrence of psoriasiform dermatitis is unclear but has been liked to IL-23 receptor polymorphism. Azathioprine-induced Sweet syndrome has also been described in association with Crohn disease. Oral metronidazole has been used for extraintestinal cutaneous Crohn disease ; intralesional or topical applications of potent topical corticosteroids or tacrolimus have helped clear the ulcerations of pyoderma gangrenosum. Colectomy in Crohn disease does not prevent recurrence and should be used selectively.
Sarcoidosis
Sarcoidosis is a systemic granulomatous disorder of unknown etiology with CD4+ Th1 cell and monocyte activation, leading to hypergammaglobulinemia, sarcoidal granulomas (predominantly affecting the lungs, eyes, skin, and reticuloendothelial system), and ultimately fibrosis. The condition presents most often in adults between the ages of 20 and 40 years. In pediatric patients, it is most commonly seen in adolescents between 9 and 15 years of age, in which manifestations resemble those of adult patients. In a US series of pediatric sarcoidosis, 72% to 81% among older children were African-Americans (vs. only 7% to 28% in young children).
There is no single reliable test for sarcoidosis. Because the clinical picture may be mimicked by other diseases, histologic confirmation is advisable. In typical lesions, the characteristic histopathologic finding consists of islands of large, pale-staining epithelioid cells containing few if any giant cells intermingled with histiocytes and lymphocytes. Sarcoidosis must also be distinguished from infectious granulomatous conditions, particularly mycobacterial and fungal infections, but cultures and special stains of biopsy sections are negative. Cutaneous anergy is common, but children should be tested for reactivity to tuberculin.
Sarcoidosis in Older Children and Adolescents
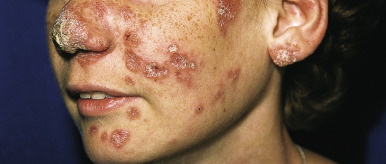
Adolescents and older children may have fever and weight loss. Cutaneous involvement occurs in 15% to 25% of pediatric patients and in older children. Several specific and nonspecific lesions have been described, although no lesion is pathognomonic. Most typical are yellow-brown to red flat-topped papules, infiltrated plaques, and nodules that show an apple-jelly color when diascopy (pressure applied with a glass slide) is performed, a characteristic sign of granulomas. Scaling is sometimes associated. These granulomatous lesions are most commonly localized to the face, and an annular configuration of lesions around the nares, lips, and eyelids is highly characteristic, although any site, including the mucosae, palms, and soles, may be involved. Lesions often occur in areas of trauma or scarification. Other cutaneous manifestations of sarcoidosis include subcutaneous, often painful nodules (Darier–Roussy sarcoidosis), ichthyosis (ichthyosiform sarcoidosis, usually most prominent on the lower extremities), generalized erythroderma with scaling, and hypopigmentation. More than 20% of affected children have erythema nodosum (see Chapter 20 ), which is often manifested at onset and is a good prognostic sign that usually portends clearance of the adenopathy. The combination of erythema nodosum, bilateral hilar adenopathy, uveitis, and fever has been called Löfgren syndrome . Lupus pernio is a variant of sarcoidosis in which soft, infiltrated, violaceous plaques are located on the nose, cheeks, ears, forehead, and on the dorsal aspects of the hands, fingers, and toes ( Fig. 25-20 ). This variant can cause significant scarring and deformity and is a marker for upper respiratory involvement, including the larynx.
The lung is the most commonly involved site in older children and adolescents, as in adults. A dry, hacking cough is the most common complaint, but patients may have dyspnea and develop parenchymal lung disease. Lymphadenopathy may be generalized, but typically involves the hilar nodes and is symmetric. More than 90% show an abnormal chest X-ray at onset. Clinical evidence of liver involvement is detected in approximately one-third of patients, although more than half show granulomas when the liver is biopsied. Splenic enlargement has been associated with extensive visceral fibrosis and a poor prognosis. Granulomatous infiltration of the heart may lead to arrhythmias, and infiltration of the lung to congestive heart failure. Some patients with renal involvement will show hypercalciuria, renal stones, and ultimately renal failure from excessive production of 1,25-dihydroxyvitamin D 3 , with or without hypercalcemia; calciphylaxis has also been reported in association with end-stage renal disease, manifesting as painful violaceous plaques, retiform purpura, or subcutaneous nodules, which can progress to nonhealing ulcers and cutaneous gangrene.
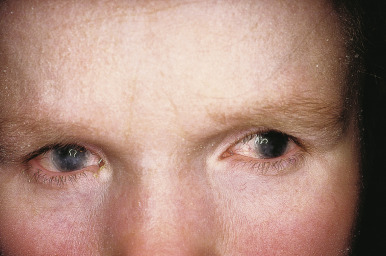
Although more common in young children, arthritis is a component of sarcoidosis in older children and can be the presenting feature in more than 10%. Overall, up to 74% of pediatric patients have eye involvement, particularly anterior segment uveitis ( Fig. 25-21 ). Conjunctival granulomas are often seen on biopsy, and lacrimal gland inflammation is not uncommon. Chorioretinitis, keratitis, and glaucoma may occur as well, resulting in blindness. Neurologic manifestations are associated with a poorer prognosis, and overall affect 5% to 10% of older patients, most commonly facial nerve paralysis. The combination of uveitis, facial nerve palsy, parotid gland enlargement, and fever has been called uveoparotid fever or Heerfordt syndrome . Lytic lesions of the distal bones rarely occur and are asymptomatic.
Thorough evaluation also includes ophthalmologic examination with slit-lamp testing, chest radiographs (with chest computed tomography [CT] scanning if needed), pulmonary function testing, electrocardiogram, and 24-hour urine calcium measurements. Many affected patients have hyperglobulinemia, especially African-Americans. Patients may show hypercalcemia (7% to 24% and usually transient), leukopenia, eosinophilia (>50% of children), and usually an increased ESR. The angiotensin-converting enzyme (ACE) level is often elevated but may be increased in other granulomatous disorders as well and has a 40% false-negative rate.
The natural course of sarcoidosis in childhood is insidious, and the condition often regresses completely after many years, especially in those with an acute onset. Chronic, progressive sarcoidosis has a poorer prognosis and rarely involutes. Features that portend a worse prognosis include chronicity; lupus pernio; symptoms lasting longer than 6 months; black race; involvement of more than three organ systems; and later stage pulmonary disease. The overall mortality rate is 1% to 5%.
Corticosteroids (1 mg/kg per day initially with tapering as possible) can suppress the acute manifestations of the disorder. However, given the hazards of prolonged systemic corticosteroid therapy and the common spontaneous resolution of sarcoidosis, the administration of systemic corticosteroids is best reserved for rapidly progressive and disfiguring skin lesions, ocular disease, and significant visceral abnormalities (persistent hypercalcemia; joint involvement; lesions of the nasal, laryngeal, and bronchial mucosa; severe, debilitating, or rapidly progressing lung disease; central nervous system lesions; persistent facial palsy; myocardial involvement; and hypersplenism). Methotrexate is the most commonly used steroid-sparing systemic agent for pediatric sarcoidosis, and MMF and TNF-α inhibitors (particularly infliximab) have also been used successfully. Ophthalmic steroid preparations may be used adjunctively for eye disease. Nonsteroidal anti-inflammatory drugs (NSAIDs) can be used for the arthritis, and hydroxychloroquine and allopurinol have been used successfully for skin lesions.
Sarcoidosis in Preschool Children/Blau Syndrome
Sarcoidosis in preschool-age children differs from that of older children, adolescents, and adults, and is characterized by polyarthritis and severe uveitis (commonly suggesting a diagnosis of juvenile idiopathic arthritis [JIA]), cutaneous manifestations, and a conspicuous absence of pulmonary abnormalities ( Table 25-5 ). The majority of young children with this presentation (also called early-onset sarcoidosis [ EOS ]) have been found to have heterozygous mutations in CARD15. As such, EOS is considered to be the sporadic form of Blau syndrome, an autosomal dominant disorder with the cutaneous granulomas and other features of EOS. Blau syndrome/EOS is associated with increased flaring and morbidity and a poorer prognosis than sarcoidosis in older children and adults. The complete classic triad (skin eruption, arthritis, and recurrent uveitis) occurs in 42% of patients with Blau syndrome, and cutaneous features are usually the first to appear. Generalized asymptomatic small erythematous papules on the trunk and extremities are most commonly seen ( Figs. 25-22 and 25-23 ), often leaving small pitted scars as sequelae. However, the cutaneous lesions have variably been described as eczematous, ichthyosiform (misdiagnosed as ichthyosis vulgaris), or lichenoid. Relapses and remission are common. Erythema nodosum is the second most common skin manifestations of Blau syndrome, and leukocytoclastic vasculitis involving skin has also been described.
| Older Children and Adolescents | Preschool-Aged Children |
|---|---|
|
|
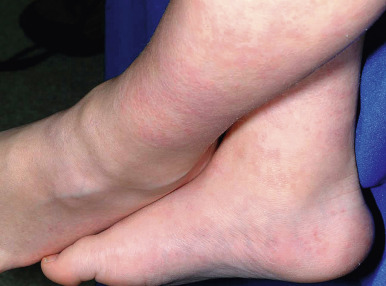
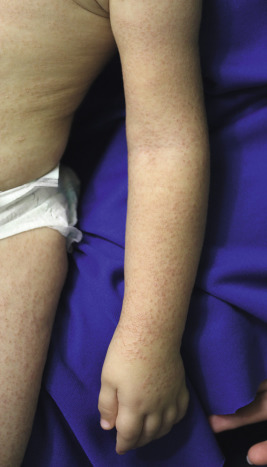
Boggy synovitis and tenosynovitis of the wrists, fingers, knees, and ankles are characteristic and show symmetric nonerosive arthritis radiographically. Despite the chronicity, joint destruction is uncommon, and range of motion is usually not reduced, in contrast to the arthritis of JIA. However, the proximal interphalangeal joints disproportionately develop contractures, leading to camptodactyly (usually congenital flexion contracture of a finger). Bone dysplasia can also be seen.
Ocular involvement is extremely common in children, especially an insidious bilateral granulomatous iridocyclitis with uveitis that involves not just the anterior uveitis (as in JIA), but also posterior uveitis. The mean age of onset of ocular disease is 4 years, and it ultimately develops in 80% of patients. Keratitis, retinitis, optic atrophy, glaucoma, cataracts, retinal detachment, and involvement of the eyelids and lacrimal glands may also occur. Of those with ocular involvement, 11% show moderate visual impairment and 16% become blind.
Other manifestations are noted in about one-third of patients. In addition to the erythema nodosum, low-grade fever, and sialadenitis with parotid gland enlargement, splenomegaly and lymphadenopathy are relatively common. Other signs are large vessel vasculitis, cranial (but not central) neuropathy, interstitial lung disease, interstitial nephritis, pericarditis, and hepatomegaly without abnormal hepatic function.
The susceptibility gene for Blau syndrome, also known as familial granulomatous arthritis, is CARD15 (also known as NOD2 ). Mutations in CARD15 lead to constitutive activation of NF-κB signaling. Mutations in CARD15/NOD2 have also been associated with the development of Crohn disease, but mutations that increase the susceptibility to Crohn disease versus Blau syndrome are in regions. The cutaneous granulomatous papules may respond to chronic administration of erythromycin but may leave tiny atrophic macules. Methotrexate and TNF-α inhibitors are most often used for extracutaneous manifestations; thalidomide and IL-1 inhibition have also been reported to be effective.
Auto-Inflammatory Disorders
Hereditary Periodic Fever Syndromes
The auto-inflammatory disorders are a group of monogenic inherited disorders characterized by intermittent or fluctuating degrees of inflammation, particularly of the abdomen, skin, and joints, without evidence of high-titer autoantibodies or antigen-specific T cells ( Table 25-6 ). One group of auto-inflammatory disorders, inflammasomopathies, results from abnormalities of the inflammasome. The inflammasome is a macromolecular complex that senses microbial products and endogenous “danger signals” to activate caspase-1 and ultimately IL-1β, key steps in the innate immune response. Some of these are intrinsic defects of the inflammasome, such as the cryopyrinopathies (defective cryopyrin leads to familial cold auto-inflammatory syndrome [FCAS], Muckle–Wells syndrome [MWS], and neonatal onset multisystemic inflammatory disorder). Others result from abnormalities or deficits in proteins that interact directly or indirectly with the inflammasome, such as pyrin (hereditary periodic fever syndromes) and PSTPIP1 (pyogenic sterile arthritis, pyoderma gangrenosum, and acne [PAPA] syndrome). The hereditary periodic fever syndromes (familial Mediterranean fever [FMF], mevalonate kinase deficiency, and TNF receptor-associated periodic syndrome) show recurrent episodes of fever, often in association with a cutaneous eruption, serositis (peritonitis, pleuritis), arthritis, and lymphadenopathy. Progressive amyloidosis of the liver and kidneys has been reported in association with several types of periodic fever syndromes and may be life-threatening.
| Disorder | Inheritance | Gene Defect | Gene Product | Associated Features |
|---|---|---|---|---|
| GRANULOMATOUS | ||||
| Blau syndrome | AD | NOD2/CARD15 | CARD15 | Onset usually <5 years old Granulomatous dermatitis, uveitis, synovitis Good response to TNF inhibitors |
| PERIODIC FEVERS | ||||
| Familial Mediterranean fever | AR | MEFV | Pyrin (marenostrin) | Recurrent fever of short duration (24–48 h) Serositis with abdominal pain, pleuritis and chest pain Pericarditis, scrotal swelling, splenomegaly, erysipelas-like eruption in minority of patients High risk of renal amyloidosis if untreated Good response to colchicine, IL-1 blockade |
| TNF receptor-associated periodic syndrome (TRAPS) | AD | TNFRSF1A | P55 TNF receptor | Recurrent fevers that are prolonged (1–3 weeks) Serositis, rash, conjunctivitis, periorbital edema, arthritis 10% to 25% incidence of renal amyloidosis Response to etanercept and IL-1 blockade |
| Mevalonate kinase deficiency (MVK) Hyper-IgD syndrome (HIDS) | AR | MVK | Mevalonate kinase | Early onset (usually first year) Periodic fever lasting 4–5 days Urticarial eruption (>90%), abdominal pain with vomiting and diarrhea, arthritis Headache, hepatosplenomegaly, painful cervical adenopathy, aphthous stomatitis, leukocytosis, high levels of IgD Response to steroids, NSAIDs, simvastatin, IL-1 blockade Often improves by adulthood Amyloidosis rare * |
| CRYOPYRINOPATHIES | ||||
| Familial cold auto-inflammatory syndrome (familial cold urticaria) | AD | ClAS1 | Cryopyrin | Cold-induced nonpruritic urticaria, arthritis, fever and chills, leukocytosis Responds to IL-1 blockade |
| Muckle–Wells syndrome | AD | CIAS1 | Cryopyrin | Recurrent urticaria, sensorineural hearing loss, amyloidosis Responds to IL-1 blockade |
| Neonatal onset multisystem disease (NOMID) † | AD | ClAS1 | Cryopyrin | Neonatal onset urticaria, chronic aseptic meningitis, arthropathy and bone deformities, fever, ocular changes and hearing loss Responds to IL-1 blockade |
| PYOGENIC | ||||
| Pyogenic sterile arthritis, pyoderma gangrenosum, and acne (PAPA) syndrome | AD | PSTPIP1/C2BP1 | PSTPIP1/C2BP1 | Pyogenic (sterile, destructive) arthritis, pyoderma gangrenosum, acne, myositis |
| Periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis (PFAPA) syndrome | Unknown | Unknown | Unknown | Periodic fevers, aphthous stomatitis, pharyngitis, and cervical adenitis Malaise, headache |
| Majeed syndrome | AR | LPIN2 | Lipin-2 | Multifocal osteomyelitis, congenital dyserythropoietic anemia, inflammatory dermatosis Responds to IL-1 blockade |
| Deficiency of the IL-1 receptor antagonist (DIRA) | AR | IL1RN | IL-1 receptor antagonist | Fetal distress, joint swelling with periosteal inflammation, severe osteopenia, lytic bone lesions, respiratory involvement, thromboses, aphthae, pyoderma gangrenosum, pustulosis Responds to IL-1 blockade |
| Deficiency of the IL-36 receptor antagonist (DITRA) | AR | IL36RN | IL-36 receptor antagonist | Usually generalized pustules, fever and malaise May have palmoplantar pustulosis or AGEP-like disorder Nail dystrophy, arthritis, cholangitis |
| CARD14-mediated pustular psoriasis (see Ch. 4 ) | AD | CARD14 | CARD14 | Pityriasis rubra pilaris, plaque psoriasis, erythroderma with pustular psoriasis Usually responds to ustekinumab May respond to methotrexate TNF inhibitor |
| Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome (see Ch. 20 ) | AR | PSMB8 | Proteasome subunit β type | Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (fever), lymphadenopathy, purpuric patches and nodules, arthralgias Same as Nakajo–Nishimura syndrome May respond to JAK inhibitor |
| STING-associated vasculopathy with onset in infancy (SAVI) syndrome | AD | TMEM173 | STING | Acral telangiectatic vasculopathy with soft tissue loss, sometimes large pustules Fevers, adenopathy, interstitial lung fibrosis May respond to JAK inhibitor |
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


