Key points
- •
The atopic march is a phenomenon characterized by progressive development of atopic dermatitis, food allergy, allergic rhinitis, and later asthma.
- •
The mechanism by which atopic dermatitis advances toward gastrointestinal or airway disease remains to be fully elucidated, but current evidence points to multiorgan epithelial breakdown contributing to pathogenesis.
- •
Initial disruption in the skin epidermal barrier permits allergen sensitization and colonization by pathogens.
- •
Allergen sensitization and pathogen colonization induce T helper 2 inflammatory response and a thymic stromal lymphopoietin-mediated pathway that further promote barrier breakdown at distant sites.
- •
Several factors contribute to epithelial permeability in the skin, gut, and lungs, including structural and junction defects, cytokine dysregulation, microbial dysbiosis, and the itch-scratch response in skin.
Introduction
Childhood onset of atopic dermatitis is often followed by sequential development of food allergy, allergic rhinitis, and later asthma, a phenomenon commonly known as atopic march ( ). While the cutaneous disease process has been the subject of extensive research, the mechanism by which atopic dermatitis progresses toward gastrointestinal or airway disease remains to be elucidated. Recent findings suggest that both inherited and acquired defects at epithelial barrier surfaces permit enhanced allergen penetration, immunoglobulin E (IgE) sensitization, and systemic T helper 2 (Th2) response ( ). In addition, levels of regulatory T cells (Tregs) in the skin, gut, and lung epithelia are diminished or ineffective at modulating the inflammatory response ( ). When the barrier of the epidermal skin surface fails, there is increased epicutaneous absorption of environmental allergens and a dysregulated immune response that predisposes to development of food allergy and asthma ( ). The initial insult to the skin may explain why atopic dermatitis is often the first disease to manifest in the atopic triad ( ). The chapter will highlight functions of the epithelial permeability barrier in the skin, gastrointestinal tract, and respiratory tract, and discuss how epithelial dysfunction linking microbiome alteration and immune dysregulation can predispose to development of atopic march.
Skin permeability
The epidermal barrier is composed of the stratum corneum and tight junctions (TJs) ( ). Epidermal barrier impairment in atopic dermatitis can result from altered lipid composition, dysfunctional and decreased structural proteins, increased skin pH, and reduced skin microbiome diversity. Cutaneous permeability defects can be assessed by measuring transepidermal water loss, which correlates with disease severity ( ; ).
Lipid alterations
Ceramides, free fatty acids, and cholesterol comprise the main stratum corneum lipids, of which ceramides are the most abundant ( ). Ceramides are a critical component of the lipid matrix that aids in preventing transepidermal water loss ( ). In atopic dermatitis there is decreased total ceramide content and alteration in ceramide chain length ( ). Specifically, short-chain ceramides are increased and long-chain ceramides are diminished, disrupting structural conformation and allowing increased transepidermal water loss ( ). Studies have shown that inflammatory cytokines decrease levels of long-chain ceramides by downregulating expression of key ceramide synthesizing enzymes such as elongases (ELOVL) and ceramide synthases (CerS), which are necessary for proper lipid formation ( ).
Similarly, free fatty acid chain length also influences conformational ordering of the lipid matrix. Long-chain fatty acids help maintain stratum corneum structure, whereas short-chain fatty acids induce less densely packed hexagonal lipid organization and disrupt the typical orthorhombic organization of the lipid lattice ( ). Lipid imbalance within atopic dermatitis lesions has been shown to be caused by downregulation of elongase (ELOVL), a key enzyme in fatty acid extension, which is often a result of systemic Th2 response ( ). Atopic dermatitis lesions generated with Th2 cytokines in human skin equivalents show reduced levels of ELOVL, indicating that Th2 response may cause changes in lipid composition in atopic dermatitis ( ).
Furthermore, sebaceous lipids are secreted onto the skin surface and function to lubricate the skin, prevent dehydration, provide antimicrobial support, and deliver antioxidants in the form of vitamin E ( ). In atopic dermatitis there is a reduction in sebum content (squalene and wax esters), suggesting an association between decreased sebaceous gland activity leading to skin barrier dysfunction and increased permeability ( ).
Filaggrin deficiency
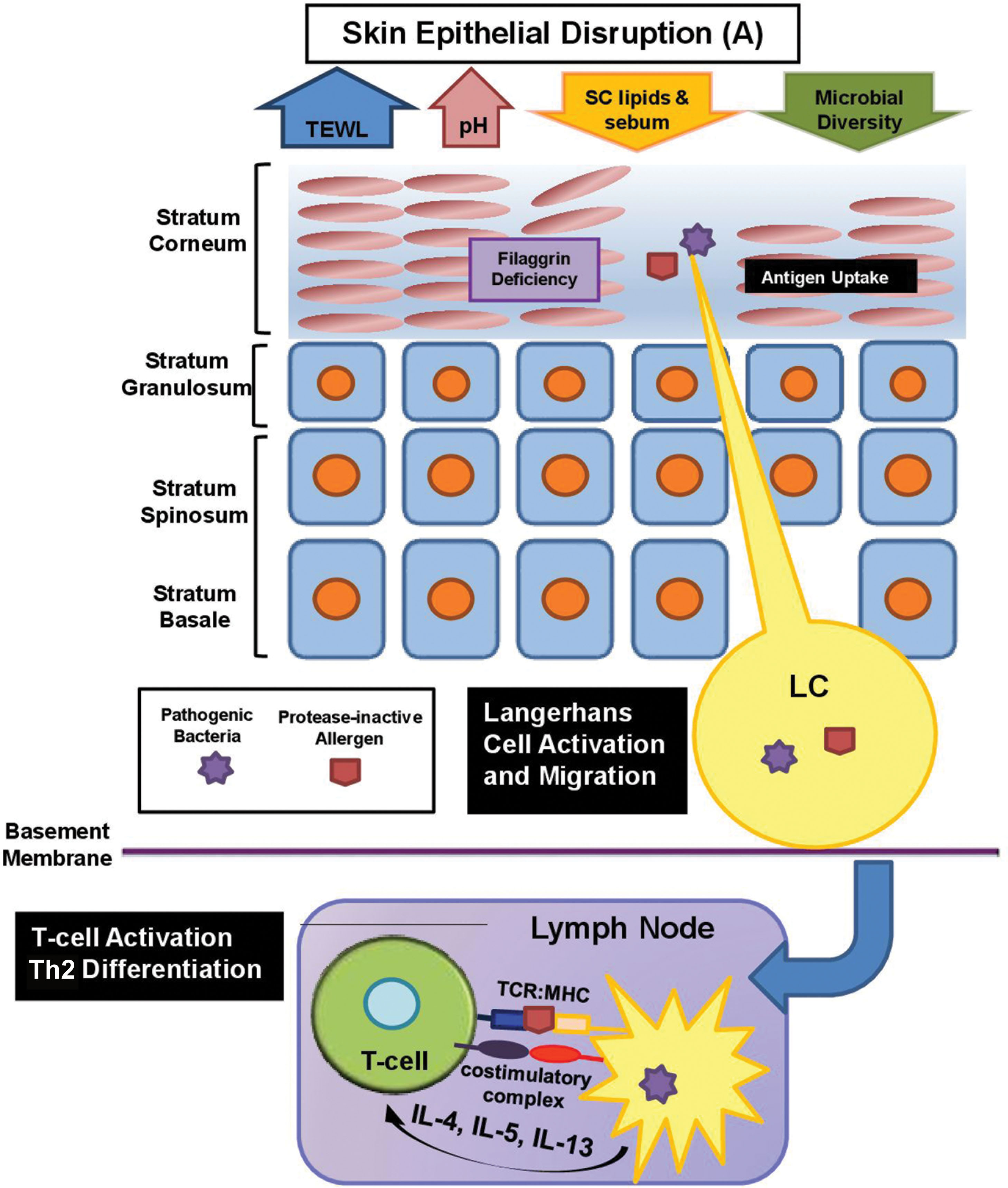
Filaggrin (FLG) is a structural protein that is fundamental in forming the cornified cell envelope and maintaining intercellular cohesion ( ). Breakdown of FLG generates alanine, pyrrolidone carboxylic acid, and urocanic acid, natural moisturizing factors that preserve hydration, lower surface pH, and contribute to skin antimicrobial defense ( ). FLG deficiency leads to increased epidermal pH, which promotes serine protease activity to degrade stratum corneum desmosomes and inhibits ceramide production ( ). Inherited loss-of-function mutation in one or both FLG alleles results in either a reduced (heterozygous) or complete absence (homozygous) of epidermal FLG ( ). In mouse models with FLG–/– genotype there is expansion of innate lymphoid cells type 2 (ILC2) in the skin and airway, leading to atopic dermatitis and pulmonary inflammation, respectively ( ). FLG expression can also be downregulated by Th2 cytokines (interleukin-4 [IL4] and IL13) and environmental factors (low humidity, ultraviolet [UV] radiation, and external irritant) ( ). Studies have shown that individuals with reduced FLG levels due to loss of function mutations exhibit early-onset atopic dermatitis that is often more persistent and closely associated with asthma and food allergy than those with normal FLG levels ( ). Fig. 15.1 illustrates physiologic alterations as a result of FLG defect.
Tight junction defects
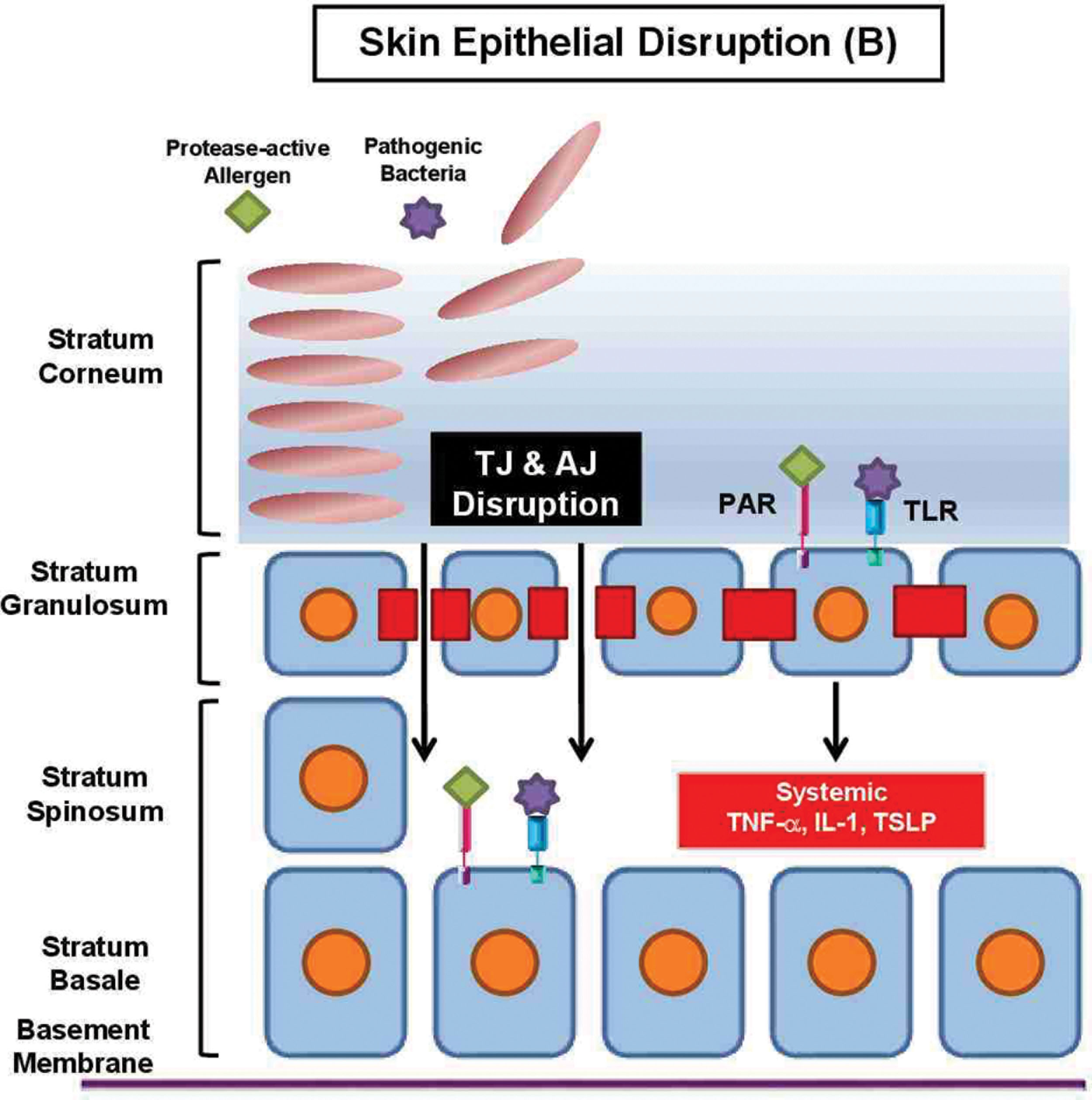
TJs are composed of transmembrane proteins—claudins, occludins, and junctional adhesion molecules—localized to the paracellular spaces of the stratum granulosum ( ). Claudin-1 and claudin-4 are responsible for intercellular sealing and formation of the liquid-liquid interface barrier within the epidermis ( ). TJ disruption weakens barrier function by altering lipid and profilaggrin processing ( ). Allergens gain rapid entry across a disrupted TJ, engage Langerhans cells, and prime the systemic inflammatory response ( Fig. 15.2 ) ( ). Fine structures of TJs are detailed in the Chapter 11 .
Proteases
Various environmental allergens, such as house dust mite, cockroach, fungi, and pollen, contain cysteine and serine proteases ( ). The proteolytic allergens can bind protease-activated receptor on keratinocytes, triggering epidermal degradation, resulting in increased permeability and Th2-mediated inflammation ( ). Reduction in serine protease inhibitors, such as serine protease inhibitor Kazal type-5, has been shown to further enhance protease-activated pathways and contribute to disease aggravation ( ).
Itch and scratch
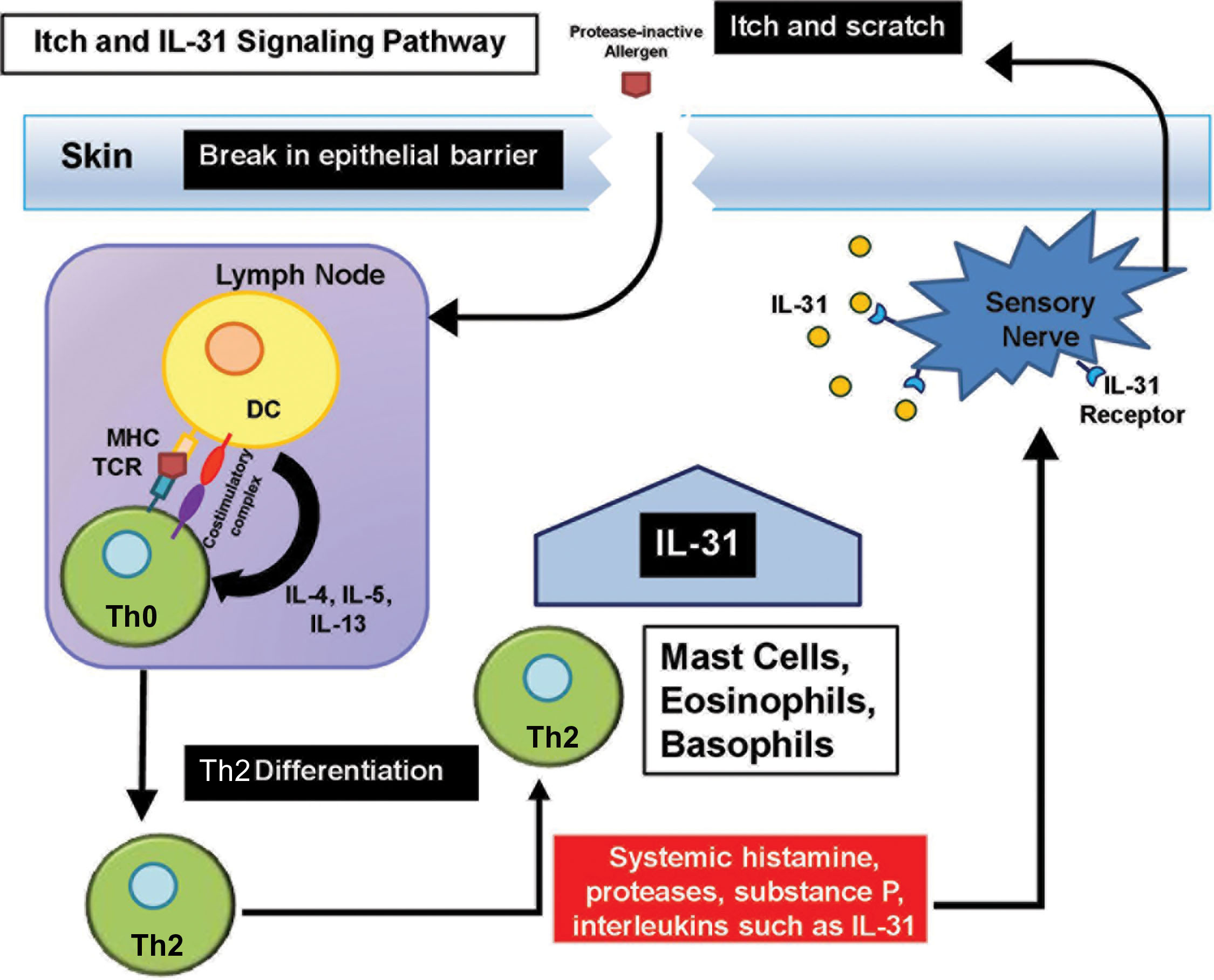
Among the many symptoms experienced by patients with atopic dermatitis, itch is nearly universal and leads to unremitting scratching that can erode the epithelial barrier ( ). Although the exact mechanism has not been elucidated, it has been purported that endogenous and exogenous triggers such as histamine, proteases, substance P, various ILs (e.g., IL31), and environmental allergens can signal through specific itch pathways (such as histamine 1 receptor, protease-activated receptor 2, neurokinin 1 receptor, IL31 receptor, and transient receptor potential cation channel ankyrin subtype 1) present on nerve fiber endings ( ).
Following breaks in the skin, allergens and pathogens gain entry across the epidermis and activate a Th2 allergic inflammatory response, which increases lymphovascular permeability allowing for the additional recruitment of pruritic initiators to the site of injury. Vigorous scratching in response to the itch can further erode the skin barrier and trigger secondary bacterial infection, perpetuating the cycle.
Recently, IL31 has emerged as a key player and potential drug target in the pathogenesis of atopic dermatitis. IL31 is primarily secreted by Th2 T cells following stimulation by IL4 and to a lesser extent by dendritic cells, mast cells, eosinophils, and basophils (see Fig. 15.2 ) ( ). IL31 primarily modulates cell-mediated immunity in the skin through perception of itch in the peripheral nervous system, in the lung through increased airway inflammation, and in the intestine through defense against microbes ( ).
Regulatory cytokines such as transforming growth factor-β (TGF-β) can downregulate IL31 levels, whereas bacteria toxins such as staphylococcal α-toxin or enterotoxin B can augment IL31 levels ( ). Selectively targeting the IL31 signaling pathway is a promising strategy to treat patients with moderate-to-severe pruritus associated with atopic dermatitis for whom conventional medical management has not been successful. Two phase II randomized, double-blind, placebo-controlled clinical trials demonstrated that monoclonal antibodies directed against the IL31 receptor can safely and effectively decrease itch and sleep disturbance, improve skin lesions, and minimize the use of topical steroids ( ). Fig. 15.3 illustrates the itch and scratch cycle and its relationship to skin barrier alteration.
Skin microbiome alteration
The skin provides an ecologic niche for a wide range of microorganisms that influence the pathogenesis of inflammatory dermatoses ( ). A diverse skin microbiota has been shown to promote normal skin homeostasis through induction of Th17 cell immune response and secretion of antimicrobial peptides that nourish commensal flora and prevent growth of pathogenic species ( ). For example, Staphylococcus epidermidis is a commensal bacterium that suppresses inflammation after skin injury and enhances innate immunity ( ). S. epidermidis also restricts growth and reproduction of pathogenic gram-positive species through competition for resources and secretion of antimicrobial peptides ( ). Conversely, Staphylococcus aureus colonization leads to skin breakdown and increased risk of atopic dermatitis development ( ). Serine proteases derived from S. aureus are capable of physically digesting the epithelial barrier, releasing enterotoxins, and inducing proinflammatory cytokines such as IL4, IL12, and IL22 ( ). Furthermore, studies show that S. aureus is found on the skin of 90% of patients with atopic dermatitis and produces ceramidases, which result in breakdown of important structural components of skin ( ). Dysbiosis, the imbalance between commensal and pathogenic microbes, is a hallmark of atopic dermatitis ( ). During atopic dermatitis flares, skin bacterial diversity is lowered, with increased S. aureus and diminished S. epidermidis ( ). Periods of remission are associated with increased bacterial diversity, with more S. epidermidis and species of Streptococcus, Corynebacterium , and Propionibacterium ( ). This temporal shift in skin microflora may be related to skin structural barrier defects that permit penetration by pathogenic species and loss of protective species ( ). Cutaneous microbiome in atopic dermatitis is further discussed in Chapter 6 .
Gut permeability
Gut epithelium forms a critical barrier that prevents entry of pathogens and noxious agents from the gastrointestinal lumen ( ). Recent studies utilizing the lactulose:mannitol ratio to measure gastrointestinal permeability have provided information on the degree of epithelial damage in individuals with atopic dermatitis ( ).
Gut barrier defects
The intestinal barrier is lined with simple columnar epithelium interspersed with goblet cells, encased in a mucous layer that functions to shield the host from digestive enzymes and microorganism penetration (see Fig. 15.3 ). TJ barrier breakdown enables dendritic cell access to luminal antigens followed by uptake in the lamina propriae ( ). In contrast to claudin-1 in the skin, claudin-2, a pore-forming claudin, is associated with increased intestinal permeability in patients with atopic dermatitis ( ). IL4/IL13 pathway dysregulation in atopic dermatitis is thought to upregulate intestinal claudin-2 expression, supporting the notion that cutaneous inflammation influences permeability defects in other organs ( ). Fig. 15.4 illustrates the disruption of gut barrier and its immunologic consequences.
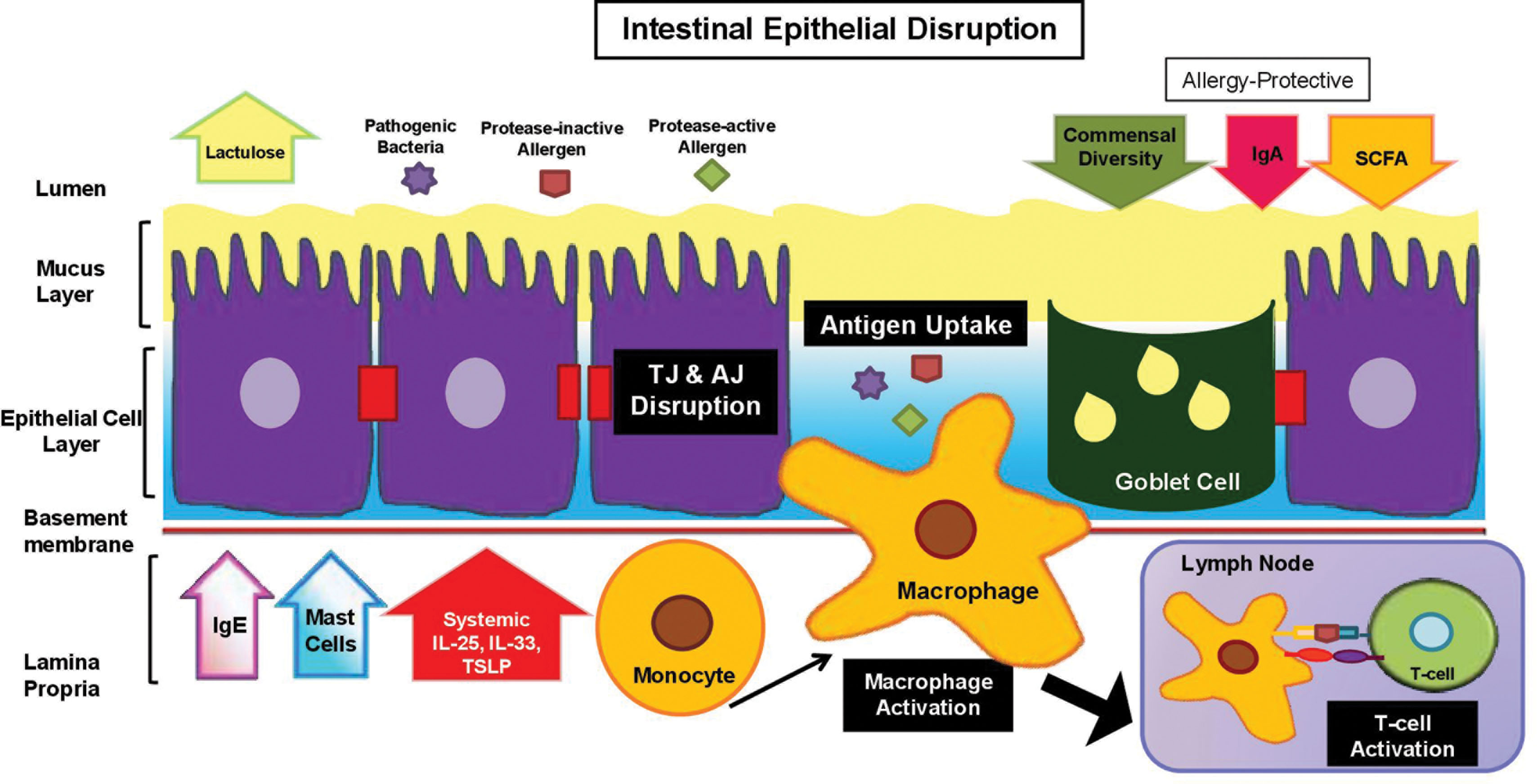
Food allergy
Atopic dermatitis is closely associated with food allergy, an abnormal immune response to common foods such as milk, eggs, peanuts, tree nuts, wheat, soy, fish, and shellfish ( ). In healthy individuals, food allergen–specific Tregs produce IL22 to maintain intestinal immune tolerance at barrier surfaces ( ). The lamina propria also contains secretory IgA that aids in immune exclusion and reduces the ability of food allergens to enter the host. In individuals with atopic dermatitis there is a depletion of allergy-protective metabolites such as bacteria-derived short-chain fatty acids and an increase in allergen danger signals such as IL25 and IL33 ( ). Early introduction of peanuts to high-risk infants (with severe eczema, egg allergy, or both) has been shown to reduce the incidence of peanut allergy significantly, which is likely to be due to dietary induction of antigen-specific Tregs ( ). These findings suggest that exposure to food allergens through a disrupted skin barrier is a risk factor for developing allergic inflammation in the gut but can be partially overcome by introduction of the allergen early in childhood.
Gut microbiome alteration
Intestinal bacterial communities evolve in response to host factors (pH, bile acids, and pancreatic enzymes), nonhost factors (nutrients, medications, and environmental allergens), and bacterial factors (toxins) ( ). In developed countries, high-fat and sugar diet and routine antibiotic use may drive atopy by diminishing microbial diversity and depleting populations of gut bacteria with barrier-protective function ( ). Early exposure to commensal gut bacteria antigens, such as bacteriocins, confers protection against atopy by restricting growth of pathogenic bacteria ( ). Patients with atopic dermatitis have a significantly lower number of intestinal commensal Bifidobacterium , which correlates with disease severity ( ). Conversely, infants with atopic dermatitis have higher numbers of pathogenic fecal Clostridium difficile ( ). In one large-scale prospective birth cohort study (KOALA), infants who developed atopic dermatitis had higher numbers of Escherichia coli and C. difficile colonization than those without atopic dermatitis ( ). E. coli and C. difficile overgrowth was associated with a decrease in beneficial bacteria, loss of immune tolerance, and increased intestinal permeability through toxin production. In contrast, a well-balanced gut microflora in infants protects against the risk of developing atopic dermatitis and later food allergy. Gut microbiome in atopic dermatitis is further discussed in Chapter 6 .
Lung permeability
The respiratory epithelium is the first site of contact with inhaled particles such as allergens, irritants, and microorganisms and is therefore essential for orchestrating appropriate inflammatory responses to eliminate foreign pathogens while limiting tissue injury ( ).
Lung epithelium defects
The respiratory tract is made of pseudostratified epithelium, comprising ciliated cells, mucus-producing goblet cells, and undifferentiated basal cells ( Fig. 15.5 ) ( ). The mucosal layer contains mucins, defensins, IgA, and antioxidants, which form an integral part of the innate immune response ( ). In the asthmatic airway, the mucociliary apparatus becomes remodeled toward increased mucin secretion and decreased ciliary function, resulting in mucous plugging and ultimately airway obstruction ( ). Respiratory epithelium reprogramming toward a hypersecretory phenotype has been linked to increased activity of IL4, IL12, interferon-c, and tumor necrosis factor-α (TNF-α) ( ).
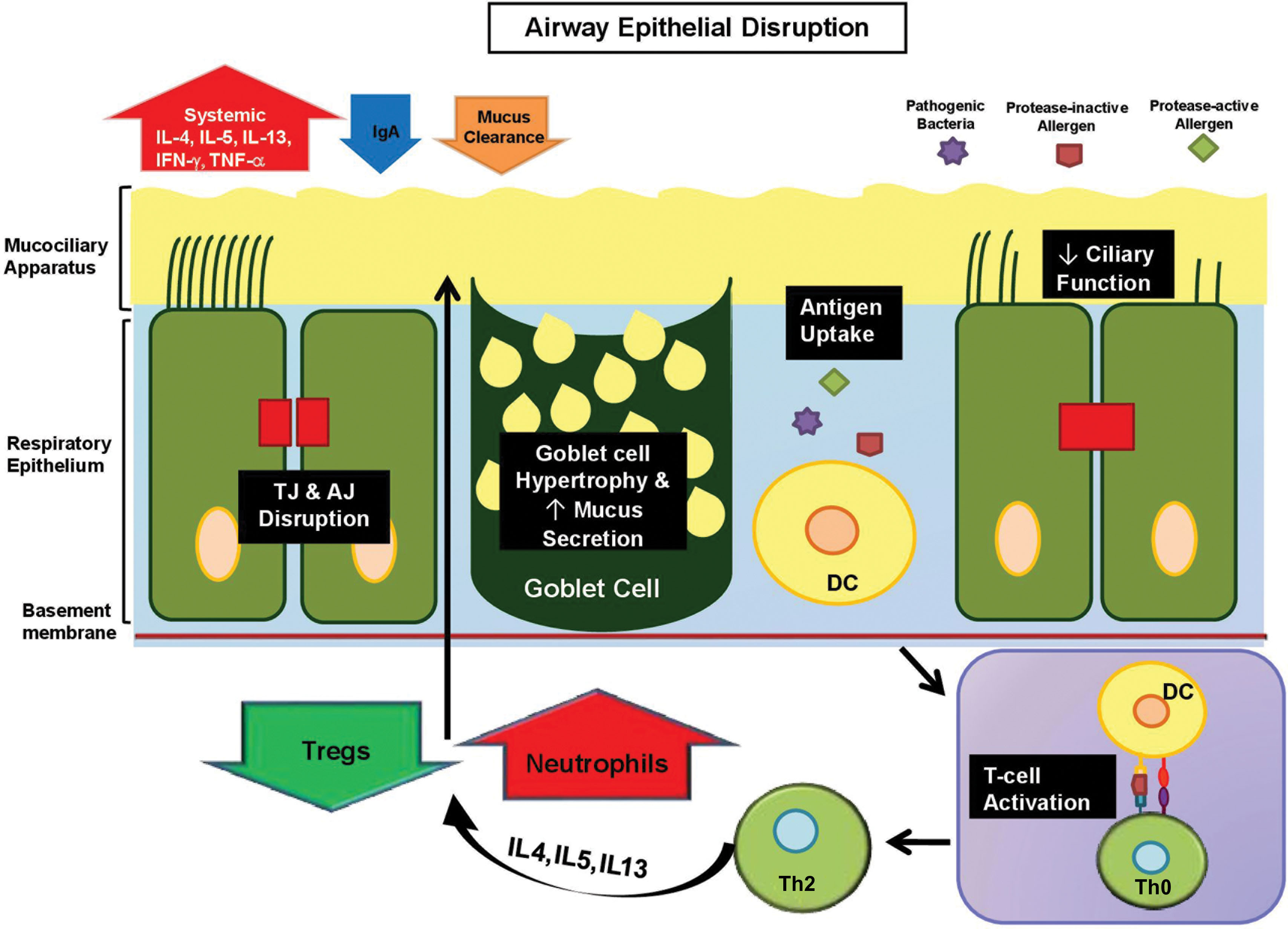
Lung epithelium junctional defects
Multiprotein subunits (claudins, occludin, zona occludens [ZOs], and junctional adhesion molecules) comprise the respiratory TJs that regulate permeability, promote cell-cell adhesion, and maintain barrier integrity ( ). In asthmatic airways, claudin-1, claudin-4, and claudin-18 are suppressed by the IL13/Th2 pathway, which results in increased paracellular leakiness and respiratory hyperresponsiveness ( ). Occludins and ZOs, particularly ZO-1, are reduced in upper airway epithelial cells, permitting neutrophil activation and cytotoxic cytokine release, causing local tissue damage ( ). Junctional adhesion molecule abnormalities enhance viral particle binding and uptake in the basolateral space ( ). Finally, epithelial junctional complexes as a whole can be disrupted, leading to inhalation of proteolytic substances, including air pollutants, cigarette smoke, and toxins ( ).
Below the TJ are adherens junctions (AJs), composed of cadherins and catenins, which are important for epithelial adhesion, wound repair, gene regulation, and cell differentiation ( ). Internal forces, such as IL4, IL13, and histamine, directly inhibit surface expression of E-cadherin and b-catenin, while external forces, such as house dust mite, indirectly downregulate E-cadherin expression ( ). In addition, chronic inflammation downregulates caveolin-1, a stabilizer of AJs, reducing E-cadherin and b-catenin expression ( ). These findings suggest a vicious cycle operating in asthmatic airways, in which barrier dysfunction leads to allergen sensitization, followed by Th2 polarization and secretion of cytokines that further perpetrate airway epithelial leakiness.
Airway microbiome alteration
The neonatal period represents the most vulnerable window for allergen-induced allergic inflammation, a susceptibility that diminishes after the development of respiratory microbiome in the first 3 weeks of life ( ). Living in an environment with diverse microbial flora reduces the incidence of allergic disease ( ). Disturbances in microbial balance shift toward overgrowth of Haemophilus, Moraxella, Neisseria , and Streptococcus ( ). In addition, Pseudomonas and Staphylococcus are frequent colonizers of sinus tracts in patients with chronic rhinosinusitis, which predisposes to the development of asthma ( ). Upon pathogen entry, a systemic inflammatory response upregulates TNF, IL1, IL6, and IL8, which have been shown to promote epithelial barrier dysfunction by decreasing expression of occludin, claudins, E-cadherin, and catenin ( ). Upper airway disease may also be influenced by gastrointestinal microflora. Bacterial fermentation of dietary fibers produces short-chain fatty acids that can mitigate allergic responses and improve respiratory function ( ). Microbiome-derived short-chain fatty acids, such as propionate, butyrate, and acetate, dampen the Th2 inflammatory response in the lung by directly inducing Treg upregulation and exerting epigenetic modifications in the forkhead box P3 pathway ( ). Studies examining a high-fiber diet in patients with atopy have suggested a role for short-chain fatty acids in promoting tolerance to antigen exposure in the lungs ( ).
The skin-gut-lung model
Although atopic dermatitis is fundamentally a dermatologic condition, cumulative evidence has supported a skin-gut-lung model of disease pathogenesis in the atopic march. Insight from immunologic studies has provided a significant breakthrough in understanding how defects in the skin could affect distant organ systems. Furthermore, microbial communities from various organs can modulate disease course and present opportunities for therapeutic intervention ( Table 15.1 ).
A compromised barrier promotes a hyperactive immune system
Epithelial cells sense pathogen-associated molecular patterns by expressing pattern-recognition receptors, including toll-like receptors, protease-activated receptors, and Nod-like receptors ( ). In atopic dermatitis, skin barrier dysfunction enhances allergen exposure (sensitization phase) and activates innate immune defenses. Antigen acquisition by Langerhans cells further activates adaptive immune cells ( ). In atopic dermatitis mouse models, epicutaneous sensitization with allergen promotes a local and systemic Th2-predominant response, characterized by increased IL4, IL5, IL13, and IgE ( ). The combination of papain (a proteolytic antigen) and mechanical barrier damage caused by tape stripping synergistically promote dermatitis and antigen-specific IgE response ( ). Following epicutaneous sensitization, airway challenge then leads to systemic inflammation characterized by IL33-mediated activation of ILC2, robust eosinophilia, and IgE/IgG1 production ( ). Systemic levels of Th2 cytokines (IL4, IL13, IL31) directly suppress FLG production ( ). This chronic inflammation impairs lipid synthesis, reduces junctional adhesiveness, increases pathogenic colonization, and compromises epithelial barrier function across multiple organ systems ( ). Thus epicutaneous sensitization on barrier-disrupted skin is sufficient to induce systemic Th2 allergic inflammation at distant sites, including intestinal tract and lower respiratory airways, and is believed to be the inciting event for the atopic march ( ).
Thymic stromal lymphopoietin (TSLP) has emerged as a key mediator by which epicutaneous sensitization can maintain chronic lung inflammation and intestinal food allergy ( ). TSLP is an IL7-like cytokine produced by keratinocytes following epidermal breakdown ( ). When released into the circulation, TSLP enhances maturation and proliferation of dendritic cells and B cells, stimulating the production of Th2 cytokines IL13 and IL31 ( ). In atopic dermatitis, TSLP overexpression in lesional keratinocytes promotes allergic sensitization and bronchial hyperresponsiveness ( ). Conversely, diminished TSLP levels result in attenuated Th2 response and airway inflammation ( ). Antigen-induced allergic inflammation in the gastrointestinal tract is also influenced by a TLSP-basophil pathway ( ). Epicutaneous sensitization to ovalbumin or peanut on an atopic dermatitis–like skin lesion followed by intragastric antigen challenge directly increases TSLP-elicited basophils in the skin, which leads to elevated antigen-specific serum IgE levels and the accumulation of mast cells in the intestine ( ). These findings suggest that inhibiting TSLP production in the skin can be useful in preventing or limiting allergen sensitization and halting progression of atopic march.
Another route of dysregulation involves IL17, a key tissue signaling cytokine that provides protection and regeneration of barrier organs such as the skin, lung, and gastrointestinal system ( ). Studies have shown that IL17 orchestrates protection against infections by enhancing the epithelial release of antimicrobial peptides and neutrophil chemoattractants, in addition to directly inducing claudin expression in the formation of TJs, thus ensuring mucosal barrier integrity ( ). Under natural exposure conditions, superantigens derived from S. aureus , such as staphylococcal enterotoxin B, directly induces IL17 expression that triggers keratinocyte production of human beta-defensin (HBD) ( ). However, in atopic dermatitis, IL17 is substantially impaired in the presence of type 2 cytokines ( ). The Th2 skin immune milieu contains abundant IL4 and IL13, which partially inhibits the IL17/HBD axis. This may partially explain why patients with atopic dermatitis have difficulty clearing S. aureus colonization. Finally, it is important to note that factors involved in the development of atopic dermatitis and associated disorders are likely different from those involved in the flares and/or maintenance of these disorders, although it is likely that there is an overlap. For example, TLSP modulation may help prevent disease recurrence or progression of the march, but may have more modest effects on the disease activity itself ( ).
“Chicken or the egg” conundrum
Parents of individuals with atopic dermatitis often share a common concern regarding the prognosis of their child’s skin disease: Who will go on to develop other manifestations of atopy such as asthma and/or allergic rhinitis and when? To answer this question, it’s important to recognize that atopic dermatitis is characterized clinically by a wide spectrum of phenotypes, from very mild forms of xerosis to keratosis pilaris–like follicular prominence to papular eczema to classic scaly edematous papules and plaques to the uncommon severely erythrodermic variant, each carrying a different risk for systemic involvement. In addition, the age at which an individual exhibits the first signs and symptoms of atopy may influence the probability of extracutaneous atopic disease ( ). Emerging work on phenotypic profiling of atopic dermatitis patients has examined the significance of atopic dermatitis onset and associative risk toward progressing to atopic march. A 1000-patient cohort study identified several atopic dermatitis phenotypes, including early-transient (early onset within age 2 and no further symptoms after age 4), early-persistent (onset within age 2 and persistence of symptoms until age 6), and a late phenotype (onset of atopic dermatitis after age 2) ( ). Children with early phenotypes were found to have an increased risk for developing asthma and food allergy, and the positve association was found to be stronger with the early-persistent phenotype ( ). The late phenotype was only associated with allergic rhinitis and not with asthma or food allergy ( ). A cohort study in France found that atopic dermatitis with multiple sensitizations, defined as a specific serum IgE concentration against common inhalant or food allergens, and atopic dermatitis with familial history of asthma both convey higher risks of asthma during childhood ( ). Furthermore, another study showed that early-onset atopic dermatitis with high atopy and high eosinophil levels is associated with an increased risk of childhood asthma but not of food allergy ( ).
Despite the emerging progress in assessing correlation of atopic dermatitis phenotype to susceptibility of developing asthma, food allergy, and allergic rhinitis, the complexity of atopic diseases commonly invokes the argument as to whether immunogen (medications, stress, toxins, bacteria, food particles) entry through leaky gut or airways causes systemic atopic responses that include atopic dermatitis, or whether immunogen entry through leaky skin causes systemic atopic responses that include asthma and food allergies. Recent work suggests that only a small proportion of children (4%–25%) actually follow a trajectory profile similar to that of the atopic march ( ). In addition, approximately 20% of patients do not have any evidence of IgE sensitization (based on skin prick tests), suggesting a degree of heterogeneity within the population ( ). As not all individuals show sequential disease progression from atopic dermatitis to food allergy/intolerance to asthma to allergic rhinitis, perhaps instead of a linear sequence the two processes occur concomitantly, each reciprocally influencing the other. This may explain why some children do not follow the atopic march and why some children may eventually exit out of the march. Finally, current evidence of twin and sibling studies suggests the association between atopic dermatitis and asthma and hay fever may be independent of shared early-life environmental factors, suggesting that further causal risk factors remain to be discovered despite there being a clear sequential association and plausible biologic mechanisms to explain such an association ( ).
Summary
This chapter aims to provide an overview of current evidence relating to the skin-gut-lung model of atopy. Abnormal epidermal epithelium serves as a site for allergic sensitization to antigens and colonization by bacteria. This induces a systemic Th2 response that facilitates barrier dysfunction at distant sites such as the intestinal and respiratory tracts. The typical sequence of clinical presentation is an initial atopic dermatitis that progresses to food allergy, allergic rhinitis, and later asthma. The causes of epidermal barrier abnormalities are complex and driven by a combination of structural, genetic, environmental, and immunologic factors. In addition, microbial diversity and dysbiosis influence disease severity, duration, and response to treatment. As there are no immediate cures for food allergy or asthma, the development of effective treatment for atopic dermatitis will prove to be an important strategy for prevention of the atopic march.
Further readings
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


