The dermal-epidermal basement membrane zone is an important epithelial and stromal interface, consisting of an intricately organized collection of intracellular, transmembrane, and extracellular matrix proteins. The basement membrane zone has several main functions including acting as a permeability barrier, forming an adhesive interface between epithelial cells and the underlying matrix, and controlling cellular organization and differentiation. This article identifies key molecular players of the dermal-epidermal membrane zone, and highlights recent research studies that have identified structural and functional roles of these components in the context of various blistering, neoplastic, and developmental syndromes.
The dermal-epidermal basement membrane zone (BMZ) is an important epithelial and stromal interface, consisting of an intricately organized collection of intracellular, transmembrane, and extracellular matrix proteins. The BMZ has several main functions: acting as a permeability barrier, forming an adhesive interface between epithelial cells and the underlying matrix, and controlling cell organization and differentiation.
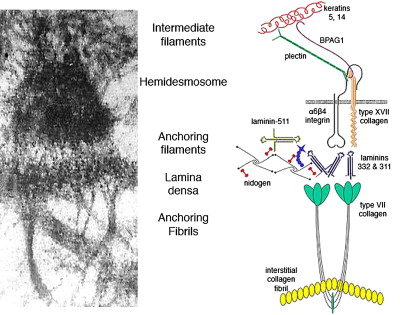
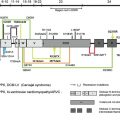
The BMZ of the skin contains a number of specialized adhesive structures, which help to promote tissue integrity in the face of disruptive external forces. At the superior aspect of the dermal-epidermal BMZ, intermediate filaments composed of keratins 5 and 14 insert on keratin linker proteins plectin and BPAG1 (BP230) as shown in Fig. 1 . BPAG1 and plectin in turn form an intricate and precise interaction with two transmembrane BMZ molecules α6β4 integrin and type XVII collagen (BP180/BPAG2). Collectively BPAG1, plectin, α6β4 integrin, and type XVII collagen comprise the electron-dense condensations of the keratinocyte plasma membrane as seen ultrastructurally by electron microscopy, which are termed “hemidesmosomes.” Jutting out below the hemidesmosome are small threadlike structures termed “anchoring filaments.” Anchoring filaments consist of or associate with type XVII collagen; α6β4 integrin; and two extracellular proteins, laminin-332 and laminin-311. These laminins assemble with other molecules including laminin-511, type IV collagen, and nidogen to form an electron-dense central structure of the BMZ termed the “lamina densa.” Finally, extending out perpendicularly as banded projections from the lamina densa are the anchoring fibrils. These structures contain polymeric associations of type VII collagen molecules, which intertwine between dermal interstitial collagen fibrils, joining the lamina densa to the papillary dermis. The dermal-epidermal BMZ serves to link the cytoskeleton of the basal epidermis, with the extensive network of interstitial collagen fibrils in the papillary dermis.
Laminins of the dermal-epidermal BMZ
The laminin family of glycoproteins is a major constituent of the BMZ, providing a critical role in providing structure to the extracellular matrix anchorage for cells and tissues. The 16 known laminin isoforms are composed of three chains (α, β, and γ) bound by disulfide bonds. To date, five α, four β, and three γ chains have been identified to create the laminin isoforms in humans. The laminin trimer resembles a cross-like structure with a large globular domain (LG domain) at the base of the cross. The LG domain is the C-terminal domain, and has been shown to function as the principal site for the interaction of laminins with various cell surface receptors. The N-terminal domain is an important mediator for the interaction of laminin with the other matrix molecules and for incorporation of laminin into the extracellular matrix.
The role of the laminin proteins has been closely linked to their interactions with integrins, heterodimeric transmembrane proteins composed of associations between α and β subunits. The specifics of laminin-integrin interaction remained largely unsolved until recently, when it was shown that the glutamic acid residue at the third position from the C-terminal region of the laminin γ chain is critically involved in the recognition of laminin by integrins. It is now also known that whereas the laminin γ chain is involved in this initial recognition, the affinity of binding between laminin and integrin is modulated by the β chain of the C terminus.
In addition to its role in the assembly of the BMZ, laminins interact with cells to influence proliferation, adhesion, and migration. To fully understand the role of laminin in pathologic states, it is important to examine the critical roles of individual laminin proteins.
Laminin-332
Laminin-332 is initially synthesized, assembled, and secreted by keratinocytes in a precursor form consisting of an α3 chain, γ2 chain, and β3 chain. Each of these chains is truncated compared with most other laminins, structurally depicted in Fig. 2 , by comparison with laminin-511, one of the other major laminins of the skin. Laminin-332 becomes even smaller after it is secreted, because it undergoes processing extracellularly at one position of its γ2 chain and at two positions of its α3 chain. The major processing enzyme is mammalian Tolloid, which is a member of the family of C-proteinases. The protein BMP-1 is another enzyme of this family that is less highly expressed in keratinocytes and fibroblasts in the skin. One of the processing sites, on the laminin α3 globular G3-4 domain, seems to have less specificity and non–C-proteinase enzymes, such as plasmin, and several MMPs has also been shown to cleave this domain. Processing of laminin-332 seems to play an important role in the skin, both during wound healing and carcinoma development.
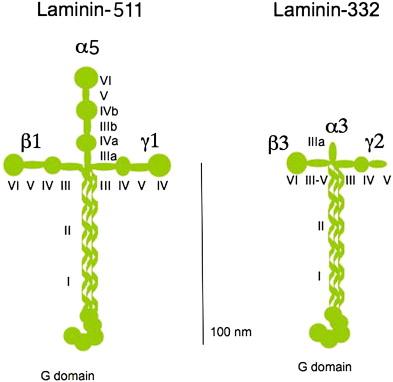
Compared with other laminins, laminin-332 has unique activity and structure. One of its unique functions is that it interacts with two major epithelial integrin receptors, α3β1 and α6β4, and promotes the formation of two separate types of attachment structures, focal adhesions and stable anchoring contacts. Laminin-332 localizes to focal adhesions through the α3β1 integrin, on which focal adhesions contribute to the migration and attachment of normal and malignant cells. It also interacts with α6β4 integrin to promote the assembly of stable anchoring contacts. Whereas focal adhesions provide short-term adhesion through reversible association of integrins, stable anchoring contacts–derived stable adhesion in epidermal cells provides increased resistance to disruptive forces. It is generally believed that focal adhesions evolve into stable anchoring contacts over time within the same area of deposited laminin-332. Structurally, laminin-332 presents unique truncations in its N-terminal regions (short arms) compared with other members of the laminin family. Despite their relatively small size, certain N-terminal domains of the short arm are important because of their ability to interact with other BMZ proteins. For example, it is known that the laminin β3 short arm domain V-III binds directly to the type VII collagen NC1 domain. It is also believed the short arm of the laminin β3 also enhances the matrix assembly of another BMZ laminin, laminin-511.
Laminin-332 is involved in various blistering diseases and skin wounds. It has been shown that during re-epithelialization after skin wounding, laminin-332 is deposited over the provisional matrix during the migration and hyperproliferation of keratinocytes. Studies using K14-Cre mice lacking the α3 subunit in the basal layer of the epidermis have shown that integrin α3β1 binds to laminin-332 that is newly deposited on the wound bed, thereby delaying keratinocyte directional migration and wound re-epithelizalization in the skin.
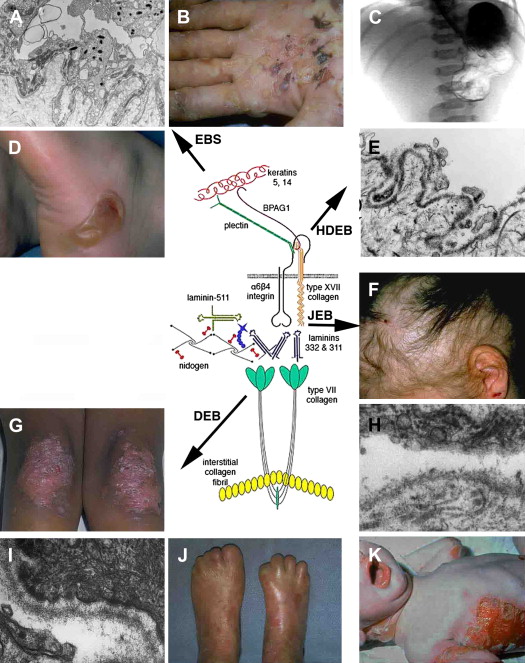
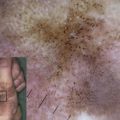
The crucial role of laminin-332 in epidermal adhesion was highlighted by the demonstration of its absence in the severe and lethal blistering Herlitz disease, junctional epidermolysis bullosa (JEB), a result of underlying laminin-332 gene mutations. JEB is just one of the many forms of epidermolysis bullosa (EB) and is characterized by intralaminar lucida blistering as depicted in Fig. 3 . JEB has largely been accepted as an untreatable genodermatosis. Recent studies have cast more light on potential therapeutic measures. Igoucheva and colleagues have demonstrated the applicability of using recombinant protein therapy for JEB by developing a protocol for production and purification of the active recombinant β3 chain that was incorporated into human keratinocytes that lack the β3 subunit. An animal model has also now been established, demonstrating the sustained phenotypic reversion of JEB in dog keratinocytes.
Most exciting are the recent results of a clinical trial of gene therapy of a non-Herlitz JEB patient. In these studies, corrective expression of laminin-332 β3 chain cDNA delivered by a retrovirus to the patient’s cells ex vivo resulted in maintenance of expression of laminin-332 and a reduction in the blistering phenotype of the localized corrected skin cell grafts even as long as 1 year after treatment. Although only performed with a single patient, these landmark studies represent the first demonstration of corrective BMZ therapy in genetic skin disease and pave the way toward future studies in this area.
Laminin-511
Laminin-511 (previously known as laminin 10) is a protein that is expressed abundantly in the BMZ underlying the interfollicular epidermis and the blood vessels in the dermis. It was first identified with the molecular cloning of the laminin α5 chain. In the first years following the initial characterization of the protein, laminin-511 remained somewhat of an enigmatic protein.
In recent years, however, advances in molecular techniques have allowed for better characterization of the structure of laminin-511 and its role in development and various skin and systemic diseases. Laminin-511 consists of three chains associated with α5, γ1, and β1 chains. It has been shown to interact with α3β1 integrin and with α-dystroglycan. The G domain of the α 5-chain is responsible for laminin-511’s interactions with both dystroglycan and α3β1 integrin. Attempts have been made to investigate further the specific epitope of the G domain that is responsible for recognition of α3β1 integrin by using a function-blocking monoclonal antibody, 4C7. The integrin binding activity did not parallel the 4C7 reactivity, however, suggesting that binding may require a strictly defined conformation of the LG domain–like 1 module, which can only be attained within an array of the LG 1 to 3 modules. Future studies need to be conducted to elucidate fully the specifics behind the interaction between laminin-511 and α3β1 integrin. It is most likely that β1 integrin and α-dystroglycan binding sites are localized to different LG modules within the laminin α5 chain G domain.
Like laminin-332, laminin-511 is involved in important skin functions. Laminin-511 is produced by human dermal microvascular endothelial cells, and highly expressed in blood vessels skin wounds. It is thought that the interaction between laminin-511 and its corresponding integrin mediates the interactions between cells and the extracellular matrix during wound angiogenesis. It has also been implicated to be an alternative adhesive ligand for promoting the proliferation and migration of epidermal keratinocytes during wound healing.
Laminin-311
Laminin-311 (previously known as laminin 6) contains the α3, β1, and γ 1 chains. It has been hypothesized that one of the unpaired cysteine residues in the laminin-311 α3 chain domain 3 EGF1 region binds to an unpaired cysteine residue in the β3 domain 6 region of laminin-332. The α3 and γ2 chains of laminin-332 are completely processed when laminin-332 is complexed with laminin-311, and these processing steps are probably necessary in the covalent association of the two laminins. Because laminins-311 and -332 share the common integrin-binding domain in the laminin α3 chain, the two have been thought to cooperatively regulate cellular functions. The mechanism of such cellular regulation is different, however, between the two proteins; laminin-311 is activated by proteolytic processing and regulates cellular adhesion and migration differently from laminin-332.
Laminins as Facilitators of Signal Transduction
The role of BMZ laminin proteins as cell-migration–promoting adhesion molecules has been widely studied and undisputed. To appreciate fully the role of laminins in the BMZ, however, it is important to appreciate the role of laminin proteins not only as facilitators of cell adhesion, but also as facilitators of critical signal transduction pathways.
It has been shown that when the γ2 chain of laminin-332’s short arm is processed by proteolysis, laminin is converted from an adhesion type to a motility type. This mobilized laminin-332 was shown to bind to syndecan-1. Such binding triggers intracellular signaling events, which negatively regulates β4 integrin, and leads to the regulation of keratinocyte adhesion and motility. In 2005, Jones and colleagues suggested that laminin-311 assembles into multimolecular fibrillar complexes with perlecan and participates in mechanical-signal transduction by a dystroglycan-dependent, integrin-dependent mechanism. These results provide new insight into the role of laminin-311 in dictating the response of epithelial cells to mechanical stimulation, carrying broad implications for the usage of mechanical ventilators in lung injury.
Finally, the dual role of laminin in promoting both cellular adhesion and signal transduction was demonstrated by the role of laminin-511 in T-cell recruitment across blood-brain barrier. In an experiment studying autoimmune encephalomyelitis, Sixt and colleagues suggested two possible roles for laminin in the endothelial BMZ: one at the level of endothelial cells resulting in reduced adhesion, and secondly at the level of T cells providing direct signals to the transmigrating cells.
Integrins of the dermal-epidermal BMZ
Integrins are a large family of heterodimeric receptor molecules comprised of transmembrane α and β subunits that are receptors for cell adhesion to the extracellular matrix or to other cells. Integrin expression is normally restricted to the basal, proliferative cell layers of the epidermis and keratinocytes.
The two main integrins of the dermal-epidermal BMZ are α6β4 and α3β1. These integrins are abundant in keratinocytes and function as cell adhesion receptors for laminin-332. Both α6β4 and α3β1 are important for maintaining the integrity of the epidermis. Ablation in mice of either of the two integrins through null mutations resulted in epidermal blistering of varying intensity. Even in the absence of α6β4 and α3β1, however, the keratinocytes retained their capacity to proliferate in the epidermis, and epidermal stratification and differentiation was normal before blister formation. These results suggested that the integrins are not essential for epidermal morphogenesis during skin development.
Despite their shared functions as receptors for laminin-332, α6β4 and α3β1 integrins are recruited to different cell adhesion structures. It is important to address the functions of the two integrins separately.
α6β4 Integrin
α6β4 Integrin is an essential component of the hemidesmosome, multiprotein adhesion complexes that promote cell-substrate adhesion in stratified and complex epithelia. It has been shown to be important in assembly of hemidesmosomes; mutating the phosphorylation of the β4 integrin, S(1424), resulted in a gradual disassembly of the hemidesmosomes. The extracellular domains of the α6β4 subunits combine together to form a ligand-binding site, whereas the intracellular domains interact with other hemidesmosomal components. The β4 integrin is only known to combine with the α6 subunit, whereas the α6 subunit can combine with either β4 or β1 subunits. As a receptor for laminin-332, α6β4 links laminin-332 anchoring filaments outside the cell with the keratin filament inside the cell. Such laminin-332 linking abilities of α6β4 integrin allow it to determine the organization of laminin-332, thereby to regulate keratinocyte adhesion, motility, and proliferation. One possible mechanism by which this happens is through β4 signaling that results in laminin-332–dependent nuclear entry of mitogen-activated protein kinases and NF-kappaB.
Deficiency or defects in α6β4 leads to epidermal diseases. Patients with mutations in the hemidesmosomal genes ITGA6 and ITGB4, which encode the α6 and β4 polypeptides, respectively, present with EB with pyloric atresia (EB-PA). The level of separation in this disease is often just above the plasma membrane, with characteristic small fragments of the basal keratinocyte plasma membrane remaining attached to the dermal side of the separation. This level of split, termed “hemidesmosomal” EB (see Fig. 3 ), is not one of the three recognized major levels of separation listed in a recent EB consensus. Nonetheless, it can be a useful feature to appreciate in making the ultrastructural diagnosis of EB. EB-PA is an autosomal-recessive disorder characterized clinically by mucocutaneous fragility and gastrointestinal atresia, which affects the pylorus. Additional complications include involvement of the urogenital tract, aplasia cutis, and failure to thrive. Prognosis of patients varies, although most affected patients die in infancy. Recent immunofluorescence analysis of villous trophoblasts reported seven previously unreported mutations in the α6β4 integrin genes in 19 pregnancy cases of PA-JEB, suggesting potential mechanisms for prenatal screening of this deadly disease.
α3β1 Integrin
In contrast to α6β4 integrin, α3β1 integrin is recruited to focal contacts in keratinocytes and other cells in culture. It thereby links the extracellular matrix to components of actin cytoskeleton. Similar to α6β4 integrin, α3β1 activates distinct signal transduction pathways, resulting in tyrosine phosphorylation of various cellular proteins, and activation of Rac 1. One unique characteristic of α3β1 is that only α3β1-dependent adhesion of laminin-332 is dependent on Rap1. This result provides evidence for a functional of camp-Epac-Rap1 pathway in cell adhesion and spreading.
α3β1 Integrin may also play a role in certain disease states. Recently, it was shown that epithelial cell α3β1 integrin links β-catenin and Smad signaling, which contribute to myofibroblast formation, and ultimately pulmonary fibrosis. It is also involved in metastasis of cancer and human development (discussed later).
Integrins of the dermal-epidermal BMZ
Integrins are a large family of heterodimeric receptor molecules comprised of transmembrane α and β subunits that are receptors for cell adhesion to the extracellular matrix or to other cells. Integrin expression is normally restricted to the basal, proliferative cell layers of the epidermis and keratinocytes.
The two main integrins of the dermal-epidermal BMZ are α6β4 and α3β1. These integrins are abundant in keratinocytes and function as cell adhesion receptors for laminin-332. Both α6β4 and α3β1 are important for maintaining the integrity of the epidermis. Ablation in mice of either of the two integrins through null mutations resulted in epidermal blistering of varying intensity. Even in the absence of α6β4 and α3β1, however, the keratinocytes retained their capacity to proliferate in the epidermis, and epidermal stratification and differentiation was normal before blister formation. These results suggested that the integrins are not essential for epidermal morphogenesis during skin development.
Despite their shared functions as receptors for laminin-332, α6β4 and α3β1 integrins are recruited to different cell adhesion structures. It is important to address the functions of the two integrins separately.
α6β4 Integrin
α6β4 Integrin is an essential component of the hemidesmosome, multiprotein adhesion complexes that promote cell-substrate adhesion in stratified and complex epithelia. It has been shown to be important in assembly of hemidesmosomes; mutating the phosphorylation of the β4 integrin, S(1424), resulted in a gradual disassembly of the hemidesmosomes. The extracellular domains of the α6β4 subunits combine together to form a ligand-binding site, whereas the intracellular domains interact with other hemidesmosomal components. The β4 integrin is only known to combine with the α6 subunit, whereas the α6 subunit can combine with either β4 or β1 subunits. As a receptor for laminin-332, α6β4 links laminin-332 anchoring filaments outside the cell with the keratin filament inside the cell. Such laminin-332 linking abilities of α6β4 integrin allow it to determine the organization of laminin-332, thereby to regulate keratinocyte adhesion, motility, and proliferation. One possible mechanism by which this happens is through β4 signaling that results in laminin-332–dependent nuclear entry of mitogen-activated protein kinases and NF-kappaB.
Deficiency or defects in α6β4 leads to epidermal diseases. Patients with mutations in the hemidesmosomal genes ITGA6 and ITGB4, which encode the α6 and β4 polypeptides, respectively, present with EB with pyloric atresia (EB-PA). The level of separation in this disease is often just above the plasma membrane, with characteristic small fragments of the basal keratinocyte plasma membrane remaining attached to the dermal side of the separation. This level of split, termed “hemidesmosomal” EB (see Fig. 3 ), is not one of the three recognized major levels of separation listed in a recent EB consensus. Nonetheless, it can be a useful feature to appreciate in making the ultrastructural diagnosis of EB. EB-PA is an autosomal-recessive disorder characterized clinically by mucocutaneous fragility and gastrointestinal atresia, which affects the pylorus. Additional complications include involvement of the urogenital tract, aplasia cutis, and failure to thrive. Prognosis of patients varies, although most affected patients die in infancy. Recent immunofluorescence analysis of villous trophoblasts reported seven previously unreported mutations in the α6β4 integrin genes in 19 pregnancy cases of PA-JEB, suggesting potential mechanisms for prenatal screening of this deadly disease.
α3β1 Integrin
In contrast to α6β4 integrin, α3β1 integrin is recruited to focal contacts in keratinocytes and other cells in culture. It thereby links the extracellular matrix to components of actin cytoskeleton. Similar to α6β4 integrin, α3β1 activates distinct signal transduction pathways, resulting in tyrosine phosphorylation of various cellular proteins, and activation of Rac 1. One unique characteristic of α3β1 is that only α3β1-dependent adhesion of laminin-332 is dependent on Rap1. This result provides evidence for a functional of camp-Epac-Rap1 pathway in cell adhesion and spreading.
α3β1 Integrin may also play a role in certain disease states. Recently, it was shown that epithelial cell α3β1 integrin links β-catenin and Smad signaling, which contribute to myofibroblast formation, and ultimately pulmonary fibrosis. It is also involved in metastasis of cancer and human development (discussed later).
Keratin linkers of the dermal-epidermal BMZ
Bullous Pemphigoid Antigen 1 (BPAG1/BP230)
Bullous pemphigoid antigen I (BPAG1) is a component of hemidesmosomes, multiprotein adhesion complexes that promote cell-substrate adhesion in stratified and complex epithelia. Molecular biologic studies indicated that the epidermal isoform of BPAG1 (BPAG1-e), a 230-kd protein, has cytoskeletal linker properties. The amino (N) terminal head domain is homologous to that of other plakin family members (hence called the “plakin domain”), and the carboxy (C) terminus consists of two homologous repeats and has been proposed to be an intermediate filament–binding domain. BPAG1 localizes to the inner plate on the cytoplasmic surface of the hemidesmosome and functions in the connection between hemidesmosomes and intermediate filaments. In 2000, a yeast two-hybrid screen was performed to identify the specific proteins that interact with the N terminus of human BPAG1-e. This yeast hybrid screen uncovered a protein belonging to the LAP/LERP protein family with 16 N-terminal leucine-rich repeats and a C-terminal PDZ domain. This protein, ERBIN, was shown to interact with not only BPAG1-e, but also with the C-terminus of the cytoplasmic domain of integrin β4 subunit. ERBIN was subsequently shown to be expressed in keratinocytes.
In a study by Guo and colleagues, it was shown that in BPAG1 negative transgenic mice that lacked the connection between hemidesmosomes and intermediate filaments, neither hemidesmosome stability nor cell substratum adhesion was weakened. BPAG1-e does not seem vital for hemidesmosome or BMZ assembly. Nonetheless, BPAG1-e has been shown to be involved in various diseases. It is a major protein targeted by autoantigens in patients with bullous pemphigoid, a subepidermal blistering disease first described by Lever in 1953. It has also been reported as an autoantigen in patients with paraneoplastic pemphigus, an autoimmune bullous skin disease induced by underlying malignant or benign neoplasias. Clinical symptoms of paraneoplastic pemphigus are variable, ranging from polymorphous blistering skin eruptions to severe, painful mucocutaneous ulcerations. There have been recent reports that BPAG1-e expression may be decreased in patients with dermatitis herpetiformis vulnerable to blistering, but further investigation is necessary to characterize the nature of this deficit.
Plectin
Plectin is a 450- to 500-kd cytoskeletal linker protein of 200 nm in length. It is widely distributed in a variety of stratified epithelia, muscle, and brain. In many tissues, plectin interacts with various cytoskeletal structures including actin microfilaments; intermediate filaments (keratin, desmin, vimentin); or microtubules. In skin, plectin links kertain intermediate filaments to the transmembrane collagen XVII and α6β4 integrin in the hemidesmosomes. More specifically, its interaction with integrin α6β4 is essential for the assembly and stability of hemidesmosomes. Recent investigations in the resolution of the primary α6β4-plectin complex showed that a major rearrangement of the β4 moiety follows the binding of the integrin with plectin, promoting stable adhesion or cell migration and an allosteric control of the integrin.
Patients with genetic defects in the epidermal expression of plectin form the basis of at least three disease subtypes: (1) a rare, mild form of dominant EB simplex (EBS) with mottled pigmentation ; (2) a severe recessive form of EB associated with PA and loss of skin ; and (3) a recessive form of EBS associated with muscular dystrophy. Of these diseases, the EBS associated with muscular dystrophy is the major one caused by plectin deficiency. In EBS associated with muscular dystrophy, plectin defects have been implicated to affect plasma membrane-cytoskeletal interactions in skin and muscle, thereby leading to epidermal blistering and muscle weakness. Even though classified as EBS, plectin deficiency usually produces a hemidesmosomal level of skin separation similar to that seen in the JEB subtype EP-PA described previously. When mutational analysis and immunohistochemistry were performed on EBS associated with muscular dystrophy and control skeletal muscle, a novel homozygous plectin-exon32 rod domain mutation (R2465X) was revealed.
Collagens of the dermal-epidermal BMZ
Type XVII Collagen (BPAG2/BP180)
Type XVII collagen (BPAG2 or BP180) is a 180-kd, transmembrane component of the dermal-epidermal anchoring complex, which projects beneath hemidesmosomes to mediate the adhesion of epidermal keratinocytes and other epithelial cells to the underlying BMZs. Recent molecular studies indicated that BPAG2 consists of 1532 amino acids, of which 1000 residues form a large extracellular carboxy-terminus extracellular domain, containing 15 collagenous domains. The extracellular portion of BPAG2 is constitutively shed from cell surface by ADAMs (proteinases that contain adhesive and metalloprotease domains).
BPAG2 is involved in various human diseases of the dermal-epidermal junction, in which it is either generally defective or absent. Absence of type XVII collagen has been demonstrated in a form of nonlethal JEB termed “generalized atrophic benign EB.” These patients exhibit a nonscarring alopecia (see Fig. 3 ) and extensive cutaneous blistering; however, mucosal blistering is not as pronounced as it is in the more severe JEB subtypes. Interestingly, revertant mosaicism of the COL17A1 gene occurs in some generalized atrophic benign EB patients. Autoantibodies against BPAG2 are seen in bullous pemphigoid, pemphigoid gestations mucous membrane pemphigoid, linear IgA disease, lichen planus pemphigoides, and pemphigoid nodularis.
Type VII Collagen
Type VII collagen is a large protein 426 nm in length, and is the main constituent of anchoring fibrils that are observed beneath the lamina densa. Like all collagens, type VII collagen assembles into a triple helix, and requires sufficient ascorbic acid as a cofactor for the enzyme prolyl hydroxylase to properly convert certain prolines to hydroxyproline, which is necessary for collagen molecular stability. Analysis of the deduced amino acid sequence of type VII collagen revealed the presence of a long central collagenous region characterized by repeating Gly-X-Y sequences that contain a number of noncollagenous interruptions, including a 39 amino acid noncollagenous segment in the center of the helix. Although it is initially secreted as a single triple helical molecule consisting of three chains, type VII collagen mostly exists as an antiparallel dimer, consisting of two triple helices joined by tail-to-tail carboxy-terminal overlap at the NC2 domains. The dimers then aggregate to form the anchoring fibril of 785 nm in length. The group of C-proteinases including BMP-1 and mammalian Tolloid, discussed in conjunction with laminin processing previously, also process type VII collagen in the NC2 region, which seems to facilitate antiparallel dimer formation. Type VII collagen is critical for the integrity of the epidermal-dermal junction through its ability to bind laminin-332, as discussed previously. Type VII collagen has the ability to intertwine between interstitial collagen fibrils, and to act as a purse-string to attach the lamina densa to the papillary dermis. In vitro binding studies demonstrated that a von Willebrand factor A–like motif is essential for binding of type VII collagen to collagen fibrils.
The importance of type VII collagen in maintaining the cohesion of the dermal-epidermal BMZ is demonstrated by its absence or by its functional defects because of underlying gene mutations in inherited blistering diseases collectively known as “dystrophic” EB (DEB), which is characterized by a sublamina densa level of skin separation (see Fig. 3 ). Type VII collagen defects have been shown to cause both dominant and recessive DEB, and hundreds of mutations have been identified and reported. These distinct, dystrophic forms of DEB show considerable phenotypic variability. Recently, a mutation analysis on approximately 1000 families with different forms of EB suggested a possible phenotype-genotype correlation in the dystrophic subtypes, providing the basis for more accurate genetic counseling and prenatal diagnosis for at-risk families. Additional future studies will cast more light on the use of phenotype-genotype correlations in DEB.
Many studies are being conducted not only to characterize type VII collagen defects in DEB, but also to investigate possible therapies for DEB patients. Protein-based therapies have been a topic of much investigation. There is some evidence that intradermal injection of recombinant human type VII collagen can restore collagen function in murine models. Mice receiving protein therapy showed decreased skin fragility, reduced new blister formation, and markedly prolonged survival. These promising results carry implications for the future of DEB treatment strategies. As a therapy with a more long-term corrective potential, type VII collagen gene delivery either to keratinocytes or fibroblasts has proved successful in reducing the blistering phenotype in a number of preclinical studies.
One interesting fact about patients with recessive DEB is that they often have epidermal cancers. Such predisposition arises from the fact that type VII collagen defects also lead to epidermal carcinogenesis. The relationship between type VII collagen defects and cancer is discussed later.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


