The prevalence of epidermolysis bullosa simplex (EBS) is estimated to be approximately 6 to 30 per 1 million live births. The disease is usually caused by missense mutations in KRT5 and KRT14 , encoding keratins mostly expressed in the epidermal basal layer. Major advances in understanding of the molecular basis of EBS and other keratin disorders have led to the development of DNA-based prenatal testing.
History
Although the first description of a congenital blistering disease is attributed to von Hebra in 1870, it is Hallopeau who highlighted the peculiar features of epidermolysis bullosa simplex (EBS) in 1898. For almost a century, the pathomechanisms underlying EBS remained poorly understood. Various theories were advanced, many of which revolved around a possible role for excessive release of deleterious proteases. Using electron microscopy, Anton-Lamprecht and Schnyder were the first to postulate that defective keratin function may be responsible for disease manifestations in EBS. These observations and seminal studies by Ishida-Yamamoto and colleagues ultimately led two groups of investigators to the discovery of pathogenic mutations in the genes encoding KRT5 and KRT14 in several families affected with EBS. EBS is the first keratin disorder whose genetic basis has been elucidated in humans.
Epidemiology
The prevalence of EBS is estimated to be approximately 6 to 30 per 1 million live births, although it is evident that many cases remain undiagnosed, suggesting that the actual prevalence of the disease may be higher than that measured in clinical surveys. The percentage of EBS cases relative to the total number of epidermolysis bullosa (EB) cases depends on the populations studied. For example, although in Western countries, EBS accounts for 75% to 85% of all EB cases, in Middle Eastern countries, a significantly higher percentage of dystrophic and junctional EB cases are observed, with EBS accounting for only approximately half of all EB cases.
Epidemiology
The prevalence of EBS is estimated to be approximately 6 to 30 per 1 million live births, although it is evident that many cases remain undiagnosed, suggesting that the actual prevalence of the disease may be higher than that measured in clinical surveys. The percentage of EBS cases relative to the total number of epidermolysis bullosa (EB) cases depends on the populations studied. For example, although in Western countries, EBS accounts for 75% to 85% of all EB cases, in Middle Eastern countries, a significantly higher percentage of dystrophic and junctional EB cases are observed, with EBS accounting for only approximately half of all EB cases.
Classification and common clinical features
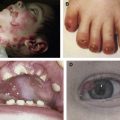
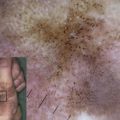
All forms of EBS manifest with blistering of the skin, usually induced by exposure to mechanical friction or trauma ( Fig. 1 A , B, and C). Palmoplantar keratoderma (thickening of the palmar and plantar skin) (see Fig. 1 E), nail dystrophy and nail shedding, alopecia, and mucosal tissue involvement are observed in rare (and often more severe) cases. Erosions and blisters heal without scarring but can leave widespread hyperpigmentation. EB nevi have been reported in all major forms of EB and may simulate clinically and dermoscopically melanoma (although no malignant transformation of these lesions has been reported and they often disappear spontaneously). Aplasia cutis congenita has also been reported in EBS.
EBS age of onset is variable, with the most severe cases manifesting at birth and less severe cases first appearing during the second or third decade of life only. Progressive improvement with age is common. High ambient temperatures and sweating are often aggravating factors.
EBS classification has undergone many revisions over the past 2 decades. The most recent classification refers to EBS as a group of inherited disorders caused by blister formation within the epidermis. As such, EBS encompasses the classical type of EBS, resulting from blister formation throughout the basal cell layer (EBS, basal type, referred to in this article as EBS), and rarer disorders associated with suprabasal blister formation (EBS, suprabasal type, referred to in this article as EBS-SB).
EBS can be classified according to its mode of inheritance. Although the overwhelming number of EBS cases in the Western world are inherited in an autosomal dominant fashion, in the Middle East, approximately 30% of the cases are caused by bilallelic recessive mutations. Another way to classify EBS is according to the anatomic distribution of the lesions. The Weber-Cockayne subtype of EBS, (designated EBS, localized) is characterized by regional involvement of the palms and soles. The Dowling-Meara subtype of EBS is at the other end of the spectrum of severity of EBS. It is characterized by a peculiar herpetiform distribution of the blisters and is often accompanied by mucosal and nail manifestations. In contrast with other forms of EBS, EBS, Dowling-Meara subtype, is associated with atrophic scarring and milia formation. A widespread but less severe form of the disease, previously termed EBS, Koebner subtype, is today classified as EBS, other generalized.
Complications
Severe skin blistering is associated with marked morbidity. Patients with EBS were found in one survey to experience a greater impairment in quality of life than individuals affected with dystrophic EB. The birth of a child with EBS can carry serious implications for interfamilial relationships. Patients report pain that can be excruciating and requires intensive treatment. Severe infections are the most common cause of mortality in EBS patients. Protein loss and involvement of the mucosae can lead to malnutrition and anemia and fluid and electrolyte imbalance. Bone mineralization defects, however, may be less common in EBS than in other forms of EB. Severe forms of EBS are associated with an increased risk for skin cancer and death by age 1.
Pathogenesis
EBS is usually caused by missense mutations in KRT5 and KRT14 , encoding keratins mostly expressed in the epidermal basal layer. Most KRT5 and KRT14 mutations have been shown to disrupt the central alpha-helical segment of these keratin molecules, thereby compromising the structure and function of the cell cytoskeleton, which becomes unable to accommodate even small amounts of mechanical stress. As a consequence of keratin cytoskeleton dysfunction, the basal cell layer is prone to fracture when exposed to friction forces. At the ultrastructural level, keratin abnormal function translates into cell vacuolization, keratin filament clumping, and blister formation, typically. Phenotype-genotype analysis revealed that mutations affecting conserved areas at the beginning and end of the central rod segment are usually associated with a more severe phenotype than mutations affecting less conserved areas of the keratin molecules, although many exceptions to this rule have been reported. In addition, not only the location but also the nature of the mutation can influence the severity of the disease.
The mechanisms leading to cell disintegration in EBS are still poorly understood. Although decreased mechanical resistance to cell deformation undeniably plays a major role in the development of cell fragility in EBS, recent data also implicate excessive apoptotic activity, possibly induced by keratin clumps, and up-regulation of the inflammatory response in the pathogenesis of the disease. In addition, some keratin mutations may affect the cytoskeletal dynamics or interfere with normal keratin post-translational modification.
Most EBS-causing mutations exert a dominant negative effect, namely, the mutant molecules interfere with the function of the normal keratins encoded by the wild-type allele. This situation has direct implications for the design of genetic therapies for EBS: introduction of a wild-type allele is unlikely to benefit EBS patients; instead, effective therapies for EBS should be aimed at eliminating the deleterious keratin molecules encoded by the mutant allele (discussed later).
EBS phenotype usually evolves over time and generally shows improvement as affected individuals get older. Down-regulation of expression of mutant keratin genes and compensatory overexpression of keratins, usually weakly expressed in the basal cell layers, such as KRT15 , have been invoked to explain this phenomenon. Somatic genetic events may also modify the course of the disease. Revertant mosaicism refers to a situation where a second mutation attenuates or abolishes the deleterious effect of the original mutation in certain areas of the skin. This phenomenon has been reported in several patients with EBS and may be more common than previously suspected. The phenotypic manifestations of EBS-causing keratin mutations can also be influenced by apparently silent sequence alterations. Finally, genetic background is also important as exemplified by the facts that phenotype-genotype correlations differ across populations and families and that keratin mutations are phenotypically expressed in a strain-dependent fashion in mice.
It seems that epidermal fragility is almost exclusively associated with defective function of the conserved central regions of keratin molecules because skin blistering is unusual in disorders resulting from mutations affecting the head or tail domain of keratins ( Fig. 2 ). For example, in Dowling-Degos disease (MIM179850), resulting from mutations in the KRT5 gene region encoding the protein head domain, blistering is not observed; in contrast, melanosome transport and epithelial growth are abnormal, resulting in reticulate hyperpigmentation of the flexures, comedo-like lesions on the neck, and pitted perioral acneiform scars. In Naegeli-Franceschetti-Jadassohn syndrome (MIM161000) and dermatopathia pigmentosa reticularis (MIM125595), which are caused by mutations affecting the head domain of KRT14, blisters are unusual; instead, patients display reticulate hyperpigmentation and lack dermatoglyphics due to deranged regulation of apoptotic activity in the basal cell layer. Despite that mutations affecting the tail domain of another keratin, KRT1, are shown to affect the process of cornification but not cell resilience, mutations affecting the KRT5 tail domain are shown to cause a dominant form of EBS, in one case indistinguishable clinically from EBS associated with mutations located in the rod-segment encoding gene region. Here, the mutation has been suggested as possibly resulting in abnormal protein folding or triggering the formation of deleterious small intracellular aggregates.
Unusual variants
Several unusual basal EBS variants deserve mention. Recessive EBS (MIM601001) can be caused by missense or nonsense mutations, resulting in loss of function rather than a dominant negative effect. EBS with mottled pigmentation (MIM131960) is characterized by skin blistering, reticulate skin pigmentation, keratoderma, and nail dystrophy (see Fig. 1 F). This subtype of EBS has been found to be strongly associated with a missense mutation (p.P25L) affecting the KRT5 head domain. p.P25L has been suggested as impairing melanin granule aggregation and keratin filament function by interfering with post-translational processing. More recent data, however, suggest that the same phenotype may also result from other mutations in KRT5 and KRT14 or from mutations in unrelated genes. EBS with migratory circinate erythema (MIM609352) is characterized by the occurrence of vesicles on the background of a migratory circinate erythema. The lesions often heal with brown pigmentation but no scarring. The disease seems to be specifically caused by a recurrent frameshift mutation affecting the structure of KRT5 tail domain. The reason for this peculiar association is elusive.
Several subtypes of basal EBS are not caused by mutations in keratins per se. For example, mutations in PLEC1 , encoding plectin, a large molecule that is part of the hemidesmosome and is known to interact with basal keratins, were found to cause a variety of EBS subtypes, including EBS with muscular dystrophy (MIM226677) ; EBS, Ogna type (MIM131950), characterized by hemorrhagic blistering ; lethal EBS ; and EBS with pyloric atresia, (MIM612138), which is also caused by mutations affecting the α6β4 integrin receptor. Mutations affecting other components of the hemidesmosomal plaque have similarly been found to cause EBS, underscoring the interdependency between hemidesmosomal junctions and epidermal cell cytoskeleton functions.
Apart from these forms of EBS associated with blister formation at the level of the basal cell layer, several relatively rare forms of EBS associated with suprabasal blistering are now recognized (EBS-SB). Ectodermal dysplasia with skin fragility (MIM604536) is an autosomal recessive disorder characterized by skin fragility, nail dystrophy, palmoplantar keratoderma, and alopecia. The disease is caused by mutations in PKF1 , encoding plakophilin 1, a component of the desmosomal plaque. Keratin intermediate filaments binding to the desmosomal plaque, at least in lower suprabasal epidermal cells, is critically dependent on normal PKF1 function, which may explain the common occurrence of blistering in EBS and in EDSF.
Lethal acantholytic EB is a new phenotype, lethal in the neonatal period, characterized by skin fragility, complete disruption of the epidermal barrier, universal alopecia, neonatal teeth, and nail loss. Histology shows suprabasal clefting and acantholysis throughout the spinous layer, mimicking pemphigus. The disease was found to result from truncation of the tail domain of desmoplakin.
EBS superficialis refers to a disease rarely reported and characterized by superficial erosions with scarring and milia formation. In one case, the disease was found to result from mutations in COL7A1 , suggesting that EBS superficialis represents a subset of dystrophic EB ( Table 1 ).
| Gene | Protein | Disease | OMIM |
|---|---|---|---|
| KRT5 | Keratin 5 | EBS, Dowling-Meara subtype | 131760 |
| EBS, Koebner subtype | 131900 | ||
| EBS, Weber-Cockayne subtype | 131800 | ||
| EBS with mottled pigmentation | 131960 | ||
| EBS with migratory circinate erythema | 609352 | ||
| Dowling-Degos disease | 179850 | ||
| K14 | Keratin 14 | EBS, Dowling-Meara subtype | 131760 |
| EBS, Koebner subtype | 131900 | ||
| EBS, Weber-Cockayne subtype | 131800 | ||
| Autosomal recessive EBS | 601001 | ||
| Dermatopathia pigmentosa reticularis | 125595 | ||
| Naegeli-Franceschetti-Jadassohn syndrome | 161000 | ||
| PLEC1 | Plectin | EBS with muscular dystrophy | 226670 |
| EBS, Ogna type | 131950 | ||
| Lethal EBS | |||
| EBS with pyloric atresia | 612138 | ||
| ITGB4 | Integrin beta 4 | Junctional EB associated with pyloric atresia | 226730 |
| EBS, Weber-Cockayne subtype | 131800 | ||
| COL17A1 | Collagen type XVII | EBS, Koebner subtype | 131900 |
| DSP | Desmoplakin | EB, lethal acantholytic | 609638 |
Diagnosis
The laboratory tools usually used to establish a diagnosis of EBS include regular histology, electron microscopy, antigen epitope mapping, immunohistochemistry, and molecular testing. Light microscopy demonstrates intraepidermal blister formation and vacuolar changes within the epidermal basal cell layer. Electron microscopy reveals blister formation throughout the basal cell layer and keratin filament clumping in Dowling-Meara cases (see Fig. 1 D). Eosinophilic inclusions are the histopathologic correlate of ultrastructural filament clumping. Antigen epitope mapping consists of staining frozen or paraffin-embedded sections with antibodies detecting proteins demarcating the level of epidermal-dermal separation, thereby determining the EB type. Antigen epitope mapping is inexpensive and, in contrast with electron microscopy, readily available at most medical institutions. Comparative studies suggest that it may perform as well as electron microscopy. Regular immunohistochemistry can be used in recessive cases to demonstrate absence of keratin intermediate filaments.
Molecular diagnosis is generally conducted in a stepwise fashion, with screening beginning by mutation analysis of KRT5 and KRT14 conserved regions; data collected in Western populations have shown that more than 40% of EBS cases are caused by a mutation affecting the KRT14 R125 residue. The existence of KRT14 pseudogenes requires the use of special techniques to avoid amplification of nonrelevant sequences. If no mutation is identified in the conserved regions of the two genes, sequence alterations are looked for in regions of the genes less often found to contain deleterious alterations. Finally, if no mutations are found in the coding regions of KRT5 and KRT14 , noncoding and nonkeratin genes, such as PLEC1 , ITGB4 , or COL17A1 , are then scrutinized. Despite intensive mutation screening, in approximately 10% to 30% of EBS cases, no mutations are found.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


