Lethal acantholytic epidermolysis bullosa (LAEB) is an autosomal recessive disorder caused by mutations in the gene encoding the desmosomal protein, desmoplakin (DSP). It is recognized as a distinct form of suprabasal epidermolysis bullosa simplex, although only a single case has been reported. The phenotype comprises severe fragility of skin and mucous membranes with marked transcutaneous fluid loss. Other features include total alopecia, neonatal teeth, and anonychia. Skin biopsy reveals abnormal desmosomes with suprabasal clefting and acantholysis and disconnection of keratin intermediate filaments from desmosomes. The DSP abnormalities present in the affected individual involved expression of truncated DSP polypeptides that lacked the tail domain of the protein. This part of DSP has a vital role in binding to keratin filaments. The affected neonate died after 10 days because of heart failure with evidence of loss of epithelial integrity in the skin, lung, gastrointestinal tract, and bladder. This article provides a clinicopathologic overview of this unique desmosomal genodermatosis, set in the context of other DSP gene mutations, both dominant and recessive, that can cause a spectrum of skin, hair, and heart abnormalities.
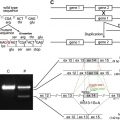
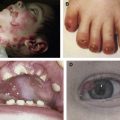
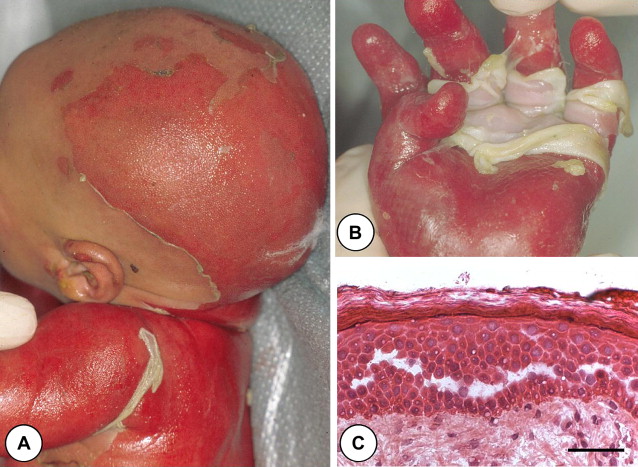
A single case of lethal acantholytic epidermolysis bullosa (LAEB) was born to nonconsanguineous parents and presented with rapidly progressive generalized epidermolysis, which started at the time of the vaginal delivery. Within 24 hours, more than two thirds of the infant’s skin was denuded, with a positive Nikolsky’s sign. There were no intact blisters or vesicles. Instead, there were areas of large sheets of detached skin with superficial red wound surfaces ( Fig. 1 A). The skin on the hands and feet had detached in a glove and stocking pattern ( Fig. 1 B). No excessive granulation tissue was noted and some sites resembled aplasia cutis. Clinical assessment also revealed complete absence of scalp hair, eyebrows, and eyelashes, although there were some discrete follicular openings on the scalp. All finger- and toenails had shed. Three triangular natal teeth were noted along with extensive erosions affecting the oral cavity. Erosions were also present on the glans penis. Over the first few days of life the skin erosions extended to more than 90% of the surface area, which led to loss of profuse amounts of fluid from the eroded skin. Ten days postpartum the child died. Autopsy revealed extensive suprabasal acantholytic separation within several epithelia, including the mouth, epiglottis, larynx, lung, gastrointestinal tract, and bladder. Cardiac dilatation was noted, but the changes in the heart were attributed to the large amounts of intravenous fluid given to compensate for the cutaneous fluid loss. The cause of death was considered to be multiorgan failure precipitated by heart failure, but there was no evidence of infection, either cutaneous or systemic. Neither parent of this infant had any cutaneous, hair, or cardiac abnormalities.
Skin pathology of lethal acantholytic epidermolysis bullosa
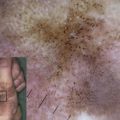
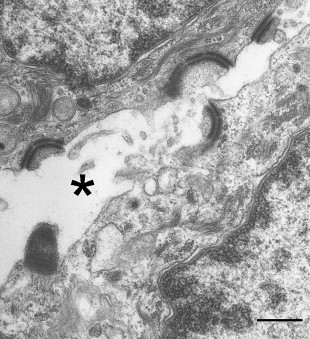
Light microscopy of skin revealed extensive suprabasal clefting with some spongiosis and acantholysis ( Fig. 1 C). These changes extended into hair follicles and eccrine ducts. Ultrastructurally, there was evidence for detachment of keratin intermediate filaments from the inner plaques of desmosomes in all layers of the epidermis ( Fig. 2 ). The number and size of the desmosomes appeared normal and both inner and outer plaques were clearly discernible. In contrast to many other autosomal-recessive blistering genodermatoses, however, immunolabeling of skin with antibodies to a panel of desmosomal and hemidesmosomal proteins was not helpful in establishing a candidate gene for mutations. Notably, all antibodies showed normal intensity staining although the pattern of staining for several desmosomal proteins was more punctate: This was the case for desmoplakin (DSP), plakoglobin, plakophilins, desmogleins, and desmocollins. In addition, labeling with keratin antibodies showed a condensed perinuclear staining, rather than a diffuse cytoplasmic pattern. The skin biopsy clue that led to the DSP gene ( DSP ) as the candidate gene was the ultrastructural observation that keratin filaments failed to connect properly to the inner dense plaques of desmosomes, a finding that had been observed previously in DSP knockout mice and in patients with nonsense/missense combinations of mutations in DSP , resulting in skin fragility–woolly hair syndrome (MIM607655).
Molecular pathology of lethal acantholytic epidermolysis bullosa
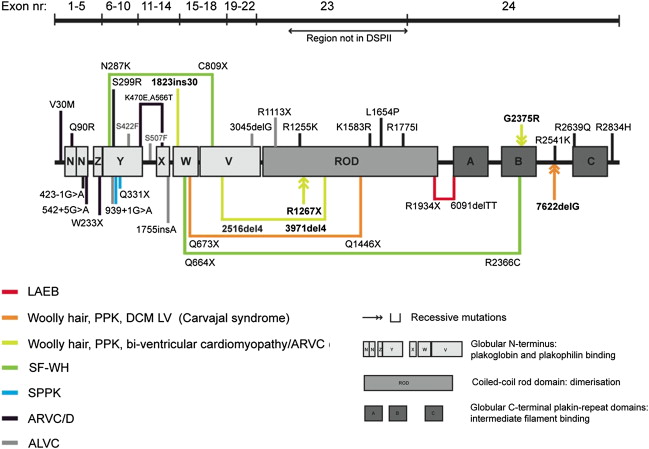
Molecular screening of DSP revealed two heterozygous mutations in the last exon (exon 24). The patient was a compound heterozygote for the mutations p.Arg1934X and a 2–base pair deletion, c.6370delTT, which led to a premature termination-codon 27 amino acids downstream ( Fig. 3 ). Nevertheless, reverse transcription–polymerase chain reaction showed that both mutant transcripts were detectable in the proband’s keratinocyte complementary DNA, indicating that neither mutation resulted in DSP haploinsufficiency by nonsense-mediated messenger RNA decay. Instead, the transcripts encoded for truncated DSP proteins (detectable by immunoblotting) that lacked the C-terminal tail. The nonsense mutation led to truncation at the end of the rod domain and thus lacked all three intermediate filament-binding subdomains (see Fig. 3 ), whereas the frameshift mutation truncated the protein shortly after the start of subdomain A and therefore lacked subdomains B and C within the keratin filament association region. These transcripts were translated into truncated proteins, confirming that the pathology of LAEB involved truncation of the DSP tail, and that the clinicopathologic abnormalities were a direct consequence of failure of intermediate filament attachment to the desmosomal inner plaques.