Pathogenic mutations have now been described in ten different desmosomal proteins: plakophilin 1 (PKP1) and 2 (PKP2); desmoplakin; plakoglobin; desmoglein 1, 2, and 4; desmocollin 2, and 3 corneodesmosin. Nevertheless, the first report of an inherited desmosomal gene disorder, published in 1997, involved loss-of-function mutations on both alleles of PKP1 , the PKP1 gene. Loss of PKP1 expression in human skin leads to skin erosions and crusting, notably with perioral fissuring as well as palmoplantar hyperkeratosis with painful cracking of the skin. Other more variable features include abnormalities of ectodermal development with growth delay, hypotrichosis or alopecia, hypohidrosis, and nail dystrophy. In contrast to some other inherited disorders of desmosomes, there is no cardiac pathology in individuals with PKP1 mutations since it is not expressed in the heart. The collection of clinical features in individuals with PKP1 mutations has been termed ectodermal dysplasia–skin fragility (ED-SF) syndrome . This genodermatosis is classified as a suprabasal form of epidermolysis bullosa simplex and thus far there have been 10 published cases. Skin biopsy shows acanthosis, acantholysis, and a reduced number of small, poorly formed desmosomes. Loss of PKP1 expression results in an integral weakness within the desmosomal plaque, leading to desmosomal detachment and cell-cell separation. Thus, the clinicopathologic features attest to the significant role of PKP1 in stabilization of desmosome structure and function, predominantly in the spinous layers of the epidermis. This article reviews the clinical, structural, and molecular pathology of this genetic disorder of desmosomes.
In 1997, details were published of a child with a clinical combination of skin fragility (erosions, fissures, scale-crust, and keratoderma) and abnormalities of ectodermal development (growth delay, hypotrichosis, and nail dystrophy). Skin biopsy showed acantholysis and loss of expression of the desmosomal protein plakophilin 1 (PKP1) and subsequently loss-of-function mutations were identified on both alleles of PKP1 , the PKP1 gene. The case was termed ectodermal dysplasia–skin fragility (ED-SF) syndrome and represented the first inherited disorder of desmosomes (MIM604536). The syndrome is now classified as a specific suprabasal form of epidermolysis bullosa simplex. Desmosomes are intricate intercellular structures that mediate adhesion between cells by anchoring intermediate filaments to the cell membrane. Structurally, desmosomes are present in certain simple, stratified, and complex epithelia, but several “desmosomal” proteins are also expressed in the nuclei of cells that do not have desmosomes. These findings indicate that desmosomal proteins may play key structural and signaling roles in several aspects of cell biology and tissue homeostasis. PKP1 is expressed throughout the epidermis, particularly in suprabasal cells, where it is required for stabilization of desmosomes. The two principal isoforms of PKP1, designated 1a and 1b, are generated through alternative splicing of exon 7. PKP1a is expressed in both desmosomes and nuclei whereas PKP1b is only expressed in nuclei. The specific biologic significance of the two isoforms, however, is unknown. The carboxyl terminus of PKP1 is required for its localization to the plasma membrane and the amino terminus is involved in the recruitment of desmoplakin to the cell membrane and desmosome assembly. PKP1 is also relevant to calcium stability of desmosomes. Following the initial report of ED-SF syndrome, nine other cases have been reported. These have established the disorder as a specific autosomal recessive genodermatosis. Pathogenic mutations in nine other desmosomal components (plakophilin 2; desmoplakin; plakoglobin; desmoglein 1, 2, and 4; desmocollin 2, and 3; and corneodesmosin) have also been reported. These mutations may cause skin, hair, or cardiac abnormalities, alone or in combination. Some inherited desmosomal disorders, notably those arising from mutations in desmoplakin or plakoglobin, may be associated with cardiomyopathy, although this is not a feature in ED-SF syndrome since PKP1 is not expressed in the heart.
Clinical features of ectodermal dysplasia–skin fragility syndrome
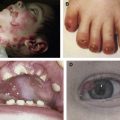
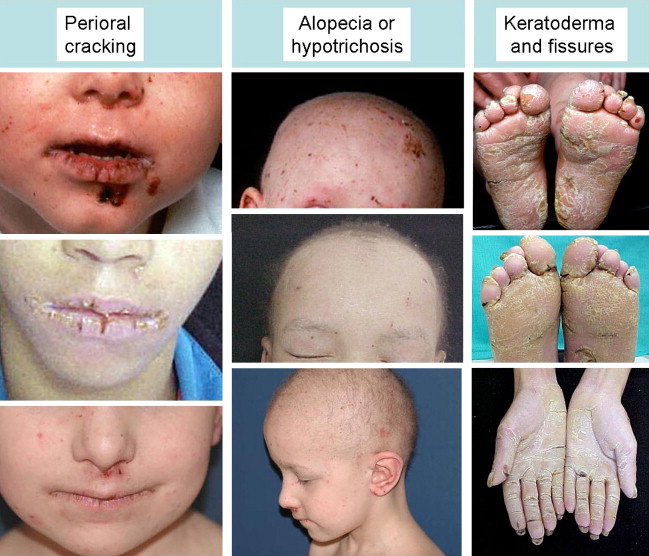
All cases of ED-SF syndrome have some degree of skin fragility, which is partly related to trauma, but which mostly occurs as spontaneous erosions and fissures. A useful clinical clue to diagnosing the syndrome is chronic cheilitis and perioral scale and cracking ( Fig. 1 ). Nearly all cases also have palmoplantar keratoderma with painful fissures that are often disabling; indeed, many affected individuals find it difficult to walk or bear weight. All cases have abnormal hair. Usually this abnormality is expressed as hypotrichosis or complete alopecia. Occasionally, depending on the consequences of the particular PKP1 mutations, the hair may be woolly rather than reduced. These hair abnormalities appear to persist over time. Nail dystrophy is a further universal clinical finding. Typically, there are growth abnormalities. Affected children are small for age, usually below the third centile for height and weight, although data regarding growth parameters into adulthood are lacking. The first case of ED-SF syndrome reported was noted to have reduced sweating, astigmatism, and dental caries. However, these do not appear to be present in most other affected individuals. Other variable clinical features include scattered scale-crust on the trunk and limbs, pruritus, recurrent systemic infections, follicular hyperkeratosis, inflammatory scaly plaques in the flexures, perianal erythema and erosions, and chronic diarrhea. One case was reported to have a patent foramen ovale, but no other cardiac pathology has been noted.
Skin pathology of ectodermal dysplasia–skin fragility syndrome
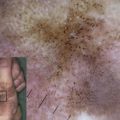
Light microscopy of skin in ED-SF syndrome typically shows hyperkeratosis and acanthosis with widening of spaces between adjacent keratinocytes, particularly throughout the spinous layer ( Fig. 2 ). Ultrastructurally, there is a reduced number of small, poorly formed desmosomes. Although there appears to be acantholysis, the actual plane of cleavage/weakness is within the desmosomal plaque (ie, inside the keratinocyte). This leads to desmosomal detachment from the keratinocyte and subsequent cell-cell separation. Immunofluorescence microscopy typically shows markedly reduced or completely absent labeling for PKP1, although the nature of the mutations in some cases may lead to some residual staining. Expression of other desmosomal proteins is usually of normal intensity, but some redistribution of staining patterns can be seen. Notably, labeling for desmoplakin tends to show more cytoplasmic staining and less membranous labeling. Likewise, keratin immunolabeling is often compacted in a perinuclear pattern with less staining at the cell periphery close to desmosomes.