Abstract
Primary immunodeficiencies represent a heterogeneous group of disorders characterized by increased susceptibility to infection. Many of these conditions have additional manifestations involving autoimmunity, allergy, lymphoproliferation, and risk of malignancy. Mucocutaneous abnormalities are often a presenting sign of primary immunodeficiencies, and recognition of these findings can facilitate diagnosis and appropriate management. Improved understanding of the genetic and molecular defects underlying many forms of immunodeficiency has led to improved treatment of affected individuals and provided important insights into immune functions.
Keywords
ataxia–telangiectasia, chronic mucocutaneous candidiasis, cartilage–hair hypoplasia syndrome, Chédiak–Higashi syndrome, complement disorders, chronic granulomatous disease, hyperimmunoglobulin E syndromes, DOCK8 deficiency, immunoglobulin deficiencies, IPEX syndrome, leukocyte adhesion deficiency, severe combined immunodeficiency (SCID), Wiskott–Aldrich syndrome
Introduction
Primary immunodeficiencies represent a heterogeneous group of inherited disorders characterized by immune system defects that result in susceptibility to infections as well as additional manifestations such as autoimmunity, allergy, and malignancy. The molecular bases have been defined for >200 monogenic primary immunodeficiency diseases, providing valuable insight into the functions of the human immune system .
Patients with genetic immunodeficiency disorders often manifest with cutaneous abnormalities . Some of these skin findings are highly characteristic of a particular disorder, while others, such as eczematous or granulomatous dermatitis, are shared by other immunodeficiencies ( Table 60.1 ). Immunodeficiency disorders covered in other chapters are listed in Table 60.2 .
| CUTANEOUS FINDINGS IN PRIMARY IMMUNODEFICIENCY DISORDERS | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Disorder | S. aureus infections | CMC | Warts | Eczematous dermatitis | Granulomatous dermatitis * (non-infectious) | LE | SVV | Ulcers (PG-like) | Other findings | |
| Superficial pyodermas | Abscesses | |||||||||
| Ataxia–telangiectasia | + | + | + | + (often ulcerate) | Oculocutaneous telangiectasias, progeric changes, CALM, BCC | |||||
| Chédiak–Higashi syndrome | + | + | + | Pigmentary dilution, hyperpigmentation in sun-exposed sites, silvery hair, bleeding diathesis, gingivitis | ||||||
| Chronic granulomatous disease | ++ | ++ | + | + | ++ (nodular, necrotic) | + | + | DLE in female carriers, Sweet syndrome, oral ulcers | ||
| Chronic mucocutaneous candidiasis | ++ | (++, but infectious due to Candida ) | Dermatophyte infections, vitiligo, alopecia areata | |||||||
| Common variable immunodeficiency | + | + | + | + | + | ++ | + | + | + | Dermatophyte infections, vitiligo, alopecia areata |
| Complement deficiencies | + | + | + | ++ | + | + | Dermatomyositis, urticaria, lipodystrophy (C3), JIA | |||
| DiGeorge syndrome † | + | + | + | |||||||
| Hyper-IgE syndromes (HIES) | ++ | ++ (cold) | + | ‡ | ++ | Neonatal papulopustular eruption | ||||
| Hyper-IgM syndrome | + | + | + | + | + | + | Oral ulcers | |||
| Idiopathic CD4 + lymphocytopenia | + | ++ | + | CD4 + T cells <300/mm 3 or <20% ^ , severe HSV infections, cryptococcal meningitis, non-tuberculous mycobacterioses, lymphoma | ||||||
| IgA deficiency | + | + | + | + | + | + | + | Vitiligo, lipodystrophia centrifugalis abdominalis | ||
| IgM deficiency | + | + | + | + | + | |||||
| IL-1 receptor-associated kinase-4 (IRAK-4) deficiency | ++ | ++ (cold) | + | |||||||
| Leukocyte adhesion deficiency | ++ (necrotic) | ++ | Poor wound healing, delayed separation of the umbilical stump, gingivitis | |||||||
| SCID | + | + | + | + | + | + * | GVHD, erythroderma; Omenn syndrome | |||
| TAP deficiency | ++ | + | + | |||||||
| WHIM syndrome | + | + | ++ | |||||||
| Wiskott–Aldrich syndrome | ++ | + | ++ | + | + | Bleeding diathesis | ||||
| X-linked agammaglobulinemia | + | ++ | + | + | Dermatomyositis-like eruption (due to echovirus); ecthyma gangrenosum | |||||
* Extensive cutaneous and extracutaneous granulomatous disease (including destructive midfacial granulomas) has been described in children with hypomorphic RAG1 or RAG2 mutations.
† Defective thymic function, hypocalcemia secondary to hypoparathyroidism, congenital heart defects, and craniofacial anomalies; due to 22q11 deletions.
‡ Patients with AR-HIES due to DOCK8 deficiency are also at increased risk of developing mucocutaneous squamous cell carcinoma and severe warts, molluscum contagiosum, and herpes simplex or varicella–zoster viral infections.
^ On more than one occasion with no evidence of HIV infection and the absence of any defined immunodeficiency or immunosuppressive therapy.
| PRIMARY IMMUNODEFICIENCIES DISCUSSED IN OTHER CHAPTERS | |
|---|---|
| Disorder(s) | Chapter |
| Autoinflammatory syndromes * | 45 |
| Netherton syndrome | 57 |
| Poikiloderma with neutropenia, Clericuzio type | 63 |
| Hypohidrotic ectodermal dysplasia with immune deficiency; immunodeficiency, myopathy, and ectodermal dysplasia | 63 |
| Hermansky–Pudlak and Griscelli syndromes | 66 |
| Dyskeratosis congenita | 67 |
| Epidermodysplasia verruciformis | 79 |
| Bloom syndrome | 87 |
| Hennekam lymphangiectasia–lymphedema syndrome | 104 |
* Characterized by immune dysregulation that often includes a component of immunodeficiency.
In general, immunodeficiency should be suspected when patients have recurrent infections of longer duration and greater severity or caused by unusual organisms. Additional signs may include incomplete clearance of infections or a poor response to antimicrobial therapy. Screening laboratory tests for a patient with recurrent cutaneous infections that raise suspicion of a primary immunodeficiency are listed in Table 60.3 Determination of the genetic etiology, which may require high-throughput approaches such as whole exome sequencing, can provide prognostic information and improve management .
| SCREENING LABORATORY TESTS TO ASSESS FOR A PRIMARY IMMUNODEFICIENCY IN A PATIENT WITH RECURRENT CUTANEOUS INFECTIONS | ||
|---|---|---|
| Test | Finding | Immunodeficiency identified |
| Complete blood count with differential, platelet count, and examination of smear |
|
|
| Hair shaft examination |
|
|
| Quantitative immunoglobulins |
|
|
| Total hemolytic complement (CH50) |
| Various complement deficiencies |
| Nitroblue tetrazolium (NBT) reduction assay |
| Chronic granulomatous disease |
| T- and B-cell analysis by flow cytometry |
| Severe combined immunodeficiency |
Ataxia–Telangiectasia
▪ Louis–Bar syndrome
- ▪
Progressive cerebellar ataxia
- ▪
Oculocutaneous telangiectasias, initially of the bulbar conjunctivae
- ▪
Selective deficiency of humoral and cell-mediated immunity, leading to sinopulmonary infections
- ▪
Increased sensitivity to ionizing radiation
- ▪
Leukemias and lymphomas
Introduction
Ataxia–telangiectasia (AT) is characterized by oculocutaneous telangiectasias, progressive cerebellar ataxia beginning in infancy, a variable immunodeficiency with a tendency to develop sinopulmonary infections, and chromosomal instability with persistent DNA damage after exposure to ionizing radiation.
Epidemiology
AT is an autosomal recessive disorder that occurs in 1 in 40 000 to 100 000 live births, with a carrier rate of up to 1% of the population.
Pathogenesis
AT results from mutations in the ataxia–telangiectasia mutated gene ( ATM ), which encodes a phosphatidylinositol 3-kinase-like serine/threonine protein kinase that plays a central role in activating apoptotic and cell cycle responses to DNA damage, particularly double-stranded breaks . These DNA breaks are sensed directly by the MRE11–RAD50–nibrin (NBN) (MRN) complex, which recruits ATM and triggers its dissociation from inactive multimers to active monomers . Autophosphorylated ATM monomers subsequently activate (via phosphorylation) a variety of targets, including p53, BRCA1, FANCD2, and Artemis in addition to NBS1 and MRE11. The result is cell cycle arrest and facilitation of DNA repair, both in the setting of external insults (e.g. ionizing radiation) and in the processing of physiologic DNA breaks that occur during V(D)J recombination in lymphocytes, telomere maintenance, and meiosis . This provides an explanation for the sensitivity to ionizing radiation, immunodeficiency, premature aging, and defective spermatogenesis that are observed in patients with AT. Progressive neurologic deterioration in affected individuals likely reflects defective DNA repair in a cell population that cannot replicate. Oxidative stress related to ATM dysfunction has also been implicated in the pathogenesis of the disease.
Of note, mutations in genes encoding components of the MRN complex cause conditions with manifestations similar to AT. MRE11 mutations underlie the AT-like disorder , which features radiosensitivity and neurologic manifestations but no telangiectasias, and NBN or rarely RAD50 mutations result in Nijmegen breakage syndrome characterized by microcephaly, immunodeficiency, chromosomal instability, and cancer predisposition.
Clinical Features
The first manifestation of AT is usually ataxia, which typically appears when the affected child begins to walk. However, the diagnosis is often not recognized until the oculocutaneous telangiectasias are well developed, at a median age of 6 years . The telangiectasias first appear at 3–6 years of age on the lateral and medial bulbar conjunctivae as red, symmetric horizontal streaks ( Fig. 60.1A ). Telangiectasias subsequently develop on the ears, eyelids, malar prominences, antecubital and popliteal fossae and presternal area, and less commonly on the dorsal aspects of the hands and feet as well as the hard and soft palate. The non-facial cutaneous telangiectasias are often subtle, resembling fine petechiae ( Fig. 60.1B ).
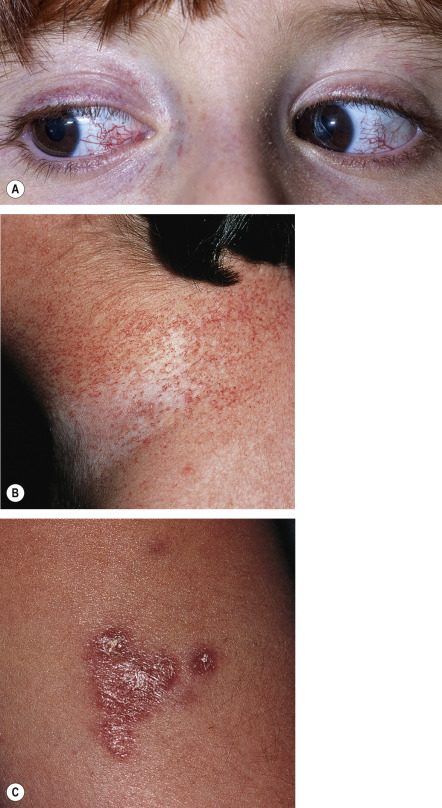
Progeric changes of the skin and hair are noted in almost 90% of patients. Subcutaneous fat is lost early and the facial skin tends to become atrophic and sclerotic. Gray hairs frequently appear in young children, and diffuse graying of the hair may occur by adolescence. Non-infectious cutaneous granulomas are another common cutaneous manifestation of AT ( Fig. 60.1C ). These granulomatous plaques tend to be persistent and ulcerate, leading to significant discomfort. Children with AT often have hyper- or hypopigmented macules as well as nevoid hyper- and/or hypopigmentation, with chromosomal instability likely explaining their predisposition to pigmentary mosaicism. Other dermatologic findings that have been described in individuals with AT include a facial papulosquamous rash, poikiloderma, hypertrichosis favoring the arms, warts, and acanthosis nigricans .
Cerebellar ataxia usually begins during infancy and is characterized by swaying of the head and trunk. Choreoathetosis, dysarthric speech, oculomotor abnormalities, and myoclonic jerks often become prominent during childhood. Despite good muscle strength, patients are typically confined to a wheelchair by 11 years of age. The facies become dull and hypotonic, with mask-like changes developing concurrently with progressive progeric features. Of note, milder AT with onset of neurologic manifestations in adulthood has been described in individuals with ATM mutations that result in residual kinase activity.
Chronic or recurrent sinopulmonary infections occur in >80% of AT patients , with the most common cause of death being bronchiectasis with respiratory failure. The majority of affected individuals also have evidence of glucose intolerance, hyperinsulinemia, and insulin resistance. Additional manifestations of AT include growth retardation (>70%), hypogonadism (especially in female patients), and intellectual disability (~30%) .
Although lymphoid malignancy has been reported as the presenting sign of AT during infancy, most neoplasias occur in young adults. Patients who survive into their late teenage years have an up to 40% risk of developing a malignancy, especially leukemia (70-fold increase compared to unaffected individuals) and lymphoma (200-fold increase). Occasionally, patients develop basal cell carcinomas (BCCs) during the third decade of life.
Carriers of heterozygous ATM mutations have a two- to threefold increase in both the risk of developing breast cancer and the likelihood of death from cancer, including malignancies of the stomach, colon, and lung as well as the breast. There is greater excess mortality in individuals <50 years of age .
Laboratory Findings
Immunologic defects in patients with AT include: (1) decreased serum levels of IgE, IgA, and IgG (especially IgG 2 and IgG 4 ) in ~80%, 70%, and 60% of patients, respectively; (2) low-molecular-weight (8S) IgM (in 80% of patients) and increased serum levels of IgM (in a minority of patients); and (3) defects in cell-mediated immunity such as lymphopenia (in 70% of patients) and deficient in vitro responses to antigens and mitogens . Affected individuals tend to have a relative deficiency of CD4 + T cells and an excess of γ/δ T cells as well as elevated serum levels of interleukin-8 (IL-8), a chemokine that may contribute to pathogenic inflammation in AT. Most patients have an absent or abnormal thymus. Individuals with no ATM activity have more severe immunologic deficits than those with low levels of ATM activity .
Spontaneous chromosomal abnormalities such as fragments, breaks, gaps, and translocations occur 2–18 times more frequently in patients with AT than in unaffected individuals, and an increased rate of telomeric shortening is also observed. Rearrangements of chromosomes 7 and 14 are particularly common and may predict the development of lymphoma. Fibroblast DNA from both patients and carriers is extremely sensitive to ionizing radiation and to radiomimetic agents such as bleomycin.
Almost all patients with AT have elevated levels of α-fetoprotein and carcinoembryonic antigen, and measuring the former can aid in establishing the diagnosis in individuals older than 2 years of age. Hepatic transaminase levels are also mildly elevated in approximately half of patients. MRI demonstrating cerebellar atrophy represents another clue to the diagnosis in patients older than 2 years. Radiosensitivity testing with the colony survival assay, analysis of radioresistant DNA synthesis (which demonstrates an abnormal S phase checkpoint), immunoblotting for the ATM protein, assessment of ATM kinase activity, karyotyping to identify 7;14 translocations, and sequencing the ATM gene may be utilized to confirm the diagnosis. Prenatal diagnosis of AT can be performed when parental mutations have been identified.
Differential Diagnosis
Children with AT may be thought to have Friedreich ataxia until the ocular telangiectasias become apparent. The conjunctival telangiectasias are occasionally misdiagnosed as conjunctivitis, but the chronicity and size of the telangiectatic vessels distinguish AT. Bloom syndrome can present with facial and occasionally conjunctival telangiectasias, café-au-lait macules, decreased immunoglobulin levels, recurrent respiratory infections, and hematologic malignancies, but patients do not have neurologic abnormalities (see Ch. 87 ). FILS syndrome – f acial dysmorphism, i mmunodeficiency, l ivedo, and s hort stature – due to polymerase ε 1 ( POLE1 ) mutations has overlapping features and can also present with telangiectasias or poikiloderma. RIDDLE syndrome is an autosomal recessive condition that features r adiosensitivity, i mmunodeficiency, facial d ysmorphism, d ifficulty le arning, abnormal motor control, and short stature due to mutations in RNF168 , which encodes a protein that mediates ubiquitin-dependent signaling at sites of DNA double-stranded breaks.
Treatment
The median lifespan in patients with AT is now >25 years. More than half of patients die from chronic sinopulmonary disease and up to a third succumb to a malignancy or complications of cancer treatment. Some of the patients who survive into adulthood show improvement in neurologic and immunologic status.
Therapy is supportive:
- •
antibiotics for infections; prophylactic antibiotics and IVIg replacement therapy for patients with severe immunodeficiency
- •
avoidance of sun exposure and use of sunscreens
- •
early physiotherapy for patients with bronchiectasis and consideration of systemic corticosteroid therapy for interstitial lung disease
- •
physical therapy to prevent contractures related to neurologic dysfunction
- •
screening for the development of malignancy.
Chronic Mucocutaneous Candidiasis
- ▪
Heterogeneous group of conditions characterized by recurrent and progressive candidal infections of the skin, nails, and mucosae
- ▪
Due to an impaired interleukin-17 response, which has a critical role in immune defense against Candida
- ▪
May be associated with additional autoimmune and infectious manifestations
Introduction
Chronic mucocutaneous candidiasis (CMC) represents a heterogeneous group of disorders characterized by progressive and recurrent infections of the skin, nails, and mucous membranes with Candida albicans . Affected individuals demonstrate an ineffective immune response against this organism, and patients with an earlier onset and greater severity of candidal infections typically have more severe underlying immunologic abnormalities.
Epidemiology
A utoimmune p olyendocrinopathy– c andidiasis– e ctodermal d ystrophy syndrome (APECED) has autosomal recessive inheritance , and other familial forms of CMC have autosomal dominant or recessive inheritance patterns ( Table 60.4 ).
| VARIANTS OF CHRONIC MUCOCUTANEOUS CANDIDIASIS (CMC) IN CHILDREN AND ADOLESCENTS | |
|---|---|
| Subgroup | Features |
| Nonsyndromic CMC (“CMC disease”) |
|
| Autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy syndrome (APECED) * |
|
| CMC due to increased STAT1 signaling (most common form) |
|
| CMC plus susceptibility to mycobacterial infections |
|
| CARD9-associated CMC |
|
| Dectin-1 deficiency |
|
| Chronic localized candidiasis (“candidal granuloma”) |
|
| Late-onset CMC |
|
| Familial chronic nail candidiasis |
|
| CMC associated with other immunodeficiency disorders |
|
| CMC associated with metabolic disorders |
|
* Ectodermal dystrophy refers primarily to dental enamel hypoplasia; also known as autoimmune polyendocrine syndrome type 1.
** Two of these three manifestations are classically required for diagnosis.
^ Mean onset at age 1.5 years; histologically, focal vacuolar interface dermatitis plus mixed superficial and deep dermal perivascular infiltrates with karyorrhexis.
Pathogenesis
Patients with CMC have an abnormal immune response to Candida spp. and sometimes to other infectious organisms. The immune defects that underlie selective predisposition to Candida infections in both APECED and non-APECED CMC patients involve an impaired response by T helper 17 (Th17) cells, which have a critical role in immune defense against Candida ( Fig. 60.2 ) .
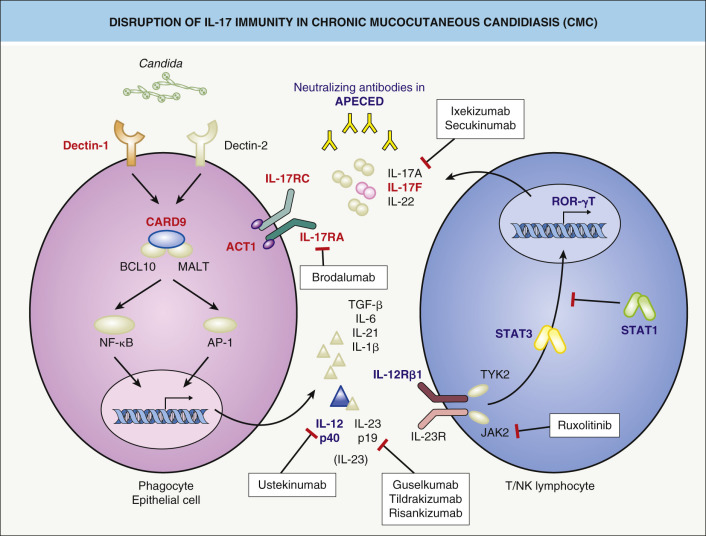
APECED results from mutations in the AIRE ( a uto i mmune re gulator) gene, which encodes a transcription factor . The AIRE protein regulates the ectopic expression of self antigens in the thymus, which allows the development of peripheral tolerance via negative selection of autoreactive T cells and the generation of antigen-specific regulatory T cells . In APECED patients, failure to delete autoreactive T cells leads to autoimmune disease . Neutralizing autoantibodies targeting Th17-associated cytokines (e.g. IL-17A/F, IL-22) have been identified in APECED patients as well as individuals with thymoma-associated CMC .
Heterozygous gain-of-function mutations in the signal transducer and activator of transcription 1 gene ( STAT1 ) have emerged as the most common cause of CMC . The resulting STAT1 activation leads to increased interferon-α/β and -γ signaling as well as repression of IL-17 production, and clinical manifestations can include predisposition to other infections, autoimmunity, and aneurysms ( Fig. 60.3 ; see Table 60.4 ). Another autosomal dominant form of CMC is caused by dominant-negative IL17F mutations that lead to IL-17F deficiency. In addition, autosomal recessive forms of CMC can result from: (1) biallelic IL17RA or IL17RC mutations that disrupt function of the IL-17 receptor; or (2) TRAF3IP2 ( ACT1 ) mutations that prevent interaction between the IL-17 receptor and its TRAF3-interacting protein 2 adaptor molecule (see Fig. 60.2 ).
CMC variants with autosomal recessive inheritance can also result from defects in the genes encoding dectin-1, a pattern-recognition receptor that binds to β-glucan in the Candida cell wall, and caspase recruitment domain family member-9 (CARD9) (see Table 60.4 ), proteins that act together to activate a Th17 response . Decreased neutrophil function is also thought to contribute to invasive fungal infections in patients with CARD9 deficiency . In other types of CMC, inadequate production of IL-23 and overproduction of IL-6, which result in an inefficient IL-17 response, have been observed.
Clinical Features
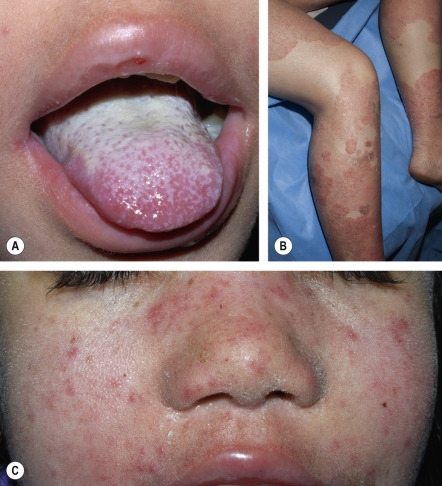
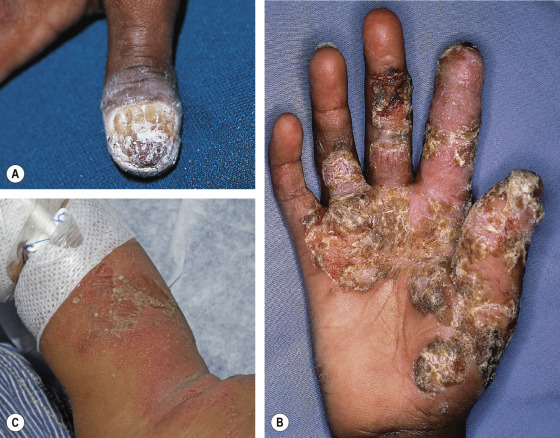
The clinical severity of CMC ranges from recurrent, treatment-resistant thrush (see Fig. 60.3A ) or a few erythematous scaly plaques and dystrophic nails to severe, generalized, crusted granulomatous plaques. The cutaneous plaques occur most frequently on the scalp and in periorificial and intertriginous sites. Scalp infections may lead to scarring alopecia. Affected nails are thickened, brittle and discolored, with associated paronychia ( Fig. 60.4 ). Although mucosal involvement is usually limited to oral thrush with hyperkeratotic plaques, chronic lesions on esophageal, genital, and laryngeal mucosae with resultant stricture formation may occur. Systemic candidiasis is rare, but cutaneous dermatophyte infections are common (see Fig. 60.3B, C ). Up to 80% of patients with childhood-onset CMC develop recurrent or severe infections with organisms other than Candida , including occasional bacterial septicemia.
Subgroups of CMC in children and adolescents include those described in Table 60.4 . An association of CMC with abnormalities of iron metabolism or (as noted above) thymoma has also been described in adult patients.
Laboratory Findings
Candidal organisms are confined to the stratum corneum and are demonstrable in scrapings and cultures. Approximately 70% of patients have direct evidence of an immunologic defect, including decreased lymphocyte proliferation in vitro , impaired cytokine production, and absent delayed-type hypersensitivity (DTH) in response to Candida , as well as nonspecific findings such as abnormal leukocyte chemotaxis or phagocytosis, depressed IgA levels, and complement dysfunction. These heterogeneous immune abnormalities reflect the variety of underlying etiologies. Candidal polysaccharides may act as serum factors that inhibit the immune response, and, in some patients, DTH to candidal antigens has been restored after antifungal therapy . Autoantibodies against type I interferons have been shown to represent a sensitive and specific marker for APECED.
Differential Diagnosis
Candidal infections, particularly thrush, are fairly common in infants. Recurrent thrush in young children with frequent otitis media may reflect alterations in bacterial flora caused by the administration of systemic antibiotics; this is seen much more often than CMC or other immunodeficiencies. Recurrent or recalcitrant candidal infections should prompt consideration of HIV infection. Clearance after traditional therapy helps to distinguish secondary candidal infections from mild CMC.
The IPEX ( i mmune dysregulation, p olyendocrinopathy, e nteropathy, X -linked) syndrome (see below) can present with autoimmune skin conditions and endocrinopathies together with recurrent infections, but the latter are typically bacterial rather than candidal. Autoimmune endocrinopathies, enteropathy, eczematous dermatitis, and a predisposition to bacterial, viral and fungal infections have also been described in patients with IL-2 receptor α-chain deficiency (see Table 60.6 ).
Treatment
Patients with CMC do not respond well to standard topical medications, and the cutaneous granulomas are especially difficult to treat. Most patients benefit from long-term therapy with systemic antifungal agents such as itraconazole and fluconazole, and alternative drugs that can be helpful in treating fluconazole-resistant Candida isolates include posaconazole, voriconazole, and echinocandins . Adjunctive therapy with granulocyte colony-stimulating factor (G-CSF) may increase IL-17 production ; nail avulsion, drainage of abscesses, and debridement of thick crusted cutaneous plaques can also be beneficial. In patients with gain-of-function STAT1 mutations, JAK inhibitors such as ruxolitinib may improve associated infectious and autoimmune disease .
Patients should be evaluated at least annually for the development of endocrinopathies, particularly if they have a diagnosis or family history of APECED or other forms of CMC with autoimmune manifestations.
Cartilage–Hair Hypoplasia Syndrome
▪ MuKusick-type metaphyseal chondrodysplasia
- ▪
Fine, sparse, hypopigmented hair
- ▪
Short-limbed dwarfism with metaphyseal dysostosis
- ▪
Doughy skin with abnormal elastic tissue
- ▪
Defective cell-mediated and (in a minority of patients) humoral immunity
Affected individuals are most likely to visit the dermatologist for their fine, sparse, hypopigmented hair, although they may also have soft, doughy skin with degenerated elastic tissue . The short-limbed dwarfism results from metaphyseal dysostosis, and ligamentous laxity is often observed. Most patients have some degree of defective cell-mediated immunity, which predisposes them to severe varicella and herpes simplex viral (HSV) infections. Approximately a third of patients also have abnormal humoral immunity , most frequently an IgA or IgG deficiency, which increases their susceptibility to recurrent respiratory tract infections that can lead to bronchiectasis. Occasionally, affected individuals present with severe combined immunodeficiency and an Omenn syndrome-like phenotype with erythroderma, eosinophilia, chronic diarrhea, lymphadenopathy, and hepatosplenomegaly (see below). Neutropenia and anemia can develop due to autoimmunity or bone marrow hypoplasia. Hirschsprung disease, other autoimmune disorders, and impaired spermatogenesis represent additional manifestations of CHH. Patients also have an increased risk of developing non-Hodgkin lymphoma and BCCs.
Hematopoietic stem cell transplantation, which can restore function of the immune system and bone marrow, should be considered in patients with severe immunodeficiency or cytopenias.
Chédiak–Higashi Syndrome
▪ Béguez César–Steinbrinck syndrome ▪ Chédiak–Higashi disease ▪ Chédiak–Steinbrinck–Higashi syndrome ▪ Chédiak–Steinbrinck anomaly
- ▪
Autosomal recessive disorder of vesicle trafficking that results in giant organelles, including melanosomes, leukocyte granules, and platelet-dense granules
- ▪
Silvery hair is accompanied by mild diffuse pigmentary dilution, often with an admixture of hyper- and hypopigmentation in sun-exposed sites; variable degree of photophobia and nystagmus
- ▪
Recurrent pyogenic infections and a mild bleeding diathesis
- ▪
An “accelerated phase” with pancytopenia and organomegaly due to lymphohistiocytic infiltration, which is fatal without hematopoietic stem cell transplantation
- ▪
Progressive neurologic deterioration in survivors
Epidemiology
Chédiak–Higashi syndrome (CHS) is a rare autosomal recessive disease reported most often in individuals of Northern European, Spanish, Middle Eastern, and Japanese ancestry . Patients of African descent have also been described .
Pathogenesis
CHS represents a disorder of vesicle trafficking and is caused by mutations in the lys osomal t rafficking regulator gene ( LYST ) . Most patients have frameshift or nonsense mutations that result in a truncated protein and lead to a severe phenotype with progression to a fatal lymphoproliferative “accelerated phase” during childhood, but 10–15% of affected individuals have missense mutations that are associated with a milder phenotype and survival to adulthood .
The giant intracytoplasmic granules that characterize CHS result from dysregulated fission/fusion of lysosome-related organelles, which include melanosomes, platelet-dense granules, and leukocyte cytolytic granules . The latter granules cannot effectively migrate or discharge their peroxidative and proteolytic enzymes, leading to defective target cell killing. The localization of cytotoxic T lymphocyte-associated antigen (CTLA-4) to enlarged vesicles rather than the cell surface may play a role in the development of lymphoproliferative disease. In addition, abnormal antigen presentation by B cells and generalized impairment in plasma membrane repair may contribute to the pathogenesis of CHS.
Clinical Features
Patients with CHS usually present in infancy with mild diffuse pigmentary dilution of the skin, hair, and eyes. In individuals with relatively dark constitutive pigmentation, areas of bronze to slate-gray hyperpigmentation may be evident in sun-exposed sites, often admixed with guttate hypopigmented macules. Hair color is variable, but a silvery, metallic sheen is characteristic. Decreased ocular pigment can result in photophobia, nystagmus, and strabismus, but visual acuity is usually normal.
Infections primarily involve the skin and respiratory tract, and they are typically caused by Staphylococcus aureus , Streptococcus pyogenes , and Streptococcus pneumoniae . The skin infections are most often superficial pyodermas. Cutaneous ulcerations resembling pyoderma gangrenosum, gingivitis, and ulceration of the oral mucosa have also been described. Although the bleeding diathesis of CHS is generally mild, some patients experience easy bruising, petechiae, and epistaxis.
Approximately 85% of patients with CHS enter a lymphoproliferative “accelerated phase” characterized by pancytopenia and lymphohistiocytic infiltration of the liver, spleen, lymph nodes, oral mucosa, and other internal organs. This “hemophagocytic syndrome” results from uncontrolled T-cell and macrophage activation, possibly associated with Epstein–Barr virus (EBV) infection, and typically leads to death by age 10 years from overwhelming infection or hemorrhage unless hematopoietic stem cell transplantation is performed.
Patients with CHS who survive the first decade of life experience progressive neurologic deterioration with abnormal gait, paresthesias, and developmental delay. Peripheral and cranial neuropathies, spinocerebellar degeneration, parkinsonism, and dementia can manifest in adult patients who were treated with hematopoietic stem cell transplantation as children as well as in those with otherwise mild disease.
Pathology
The silvery hair of patients with CHS is characterized by clumps of melanin microscopically ( Fig. 60.5 ), and giant melanosomes may be evident within melanocytes in skin biopsy specimens. A peripheral blood smear demonstrating abnormally large granules in the perinuclear area of granulocytes can assist in establishing the diagnosis. Immunologic defects in CHS include neutropenia, diminished leukocyte chemotaxis, impaired antibody-dependent cell-mediated cytotoxicity, reduced cytotoxic and regulatory T-cell activity, and markedly decreased natural killer (NK) cell function.

Differential Diagnosis
The features of CHS and the three types of Griscelli syndrome (GS), another autosomal recessive disorder that can present with immune dysfunction and silvery hair, are compared in Table 60.5 (see also Ch. 66 ). Although leukocytes generally have a normal appearance in GS, hypogammaglobulinemia can be observed. Hermansky–Pudlak syndrome (HPS) types 2, 9 (in a single patient), and 10 ( AP3B1, PLDN , and AP3D1 mutations, respectively) can result in recurrent infections due to abnormal cytotoxic T-cell function; additional manifestations include a bleeding diathesis, neurologic manifestations (in HPS10), and diffuse pigmentary dilution, but not silvery hair. Tricho-hepato-enteric syndrome (phenotypic diarrhea of infancy), an autosomal recessive disorder caused by mutations in the tetratricopeptide repeat domain 37 gene ( TTC37 ), can present with diffuse pigmentary dilution of the skin and hair, immunodeficiency, and platelet abnormalities; unlike CHS, it is also characterized by brittle hair with trichorrhexis nodosa, intractable diarrhea during infancy, primary liver disease, facial dysmorphism, and cardiac defects. Other inherited disorders characterized by lymphoproliferation due to immune dysregulation are presented in Table 60.6 .
| FEATURES OF CHÉDIAK–HIGASHI SYNDROME AND GRISCELLI SYNDROME | ||||
|---|---|---|---|---|
| CHS | GS1 * | GS2 | GS3 | |
| Gene defect | LYST | MYO5A | RAB27A | MLPH |
| Major sites of gene expression | Melanocytes, platelets, granulocytes, CNS | Melanocytes, CNS | Melanocytes, cytotoxic T cells | Melanocytes |
| Cellular defect | Vesicle trafficking (e.g. fission/fusion) | Vesicle movement/transfer | ||
| Pigmentary dilution of the skin † | + | + | + | + |
| Silvery/metallic hair | + | + | + | + |
| Hair microscopy: clumps of melanin | Small, regularly spaced | Larger, irregularly distributed | ||
| Melanocytes | Giant melanosomes | “Stuffed” with melanosomes | ||
| Neutrophils | Giant granules | Normal-appearing granules | ||
| Ocular findings | + | − | − | − |
| Bleeding diathesis | + | − | − | − |
| Recurrent infections | + | − | + | − |
| Accelerated phase | + | − | + | − |
| Primary neurologic abnormalities | + | + | − ‡ | − |
* A form of GS with manifestations limited to the skin and hair can result from deletion of the MYO5A F-exon, which is only expressed in melanocytes.
† Often accompanied by hyperpigmentation ± guttate hypopigmented macules in acral and sun-exposed sites.
‡ May develop neurologic symptoms secondary to the hemophagocytic syndrome of the accelerated phase.
| INHERITED DISORDERS CHARACTERIZED BY LYMPHOPROLIFERATION DUE TO IMMUNE DYSREGULATION | ||||
|---|---|---|---|---|
| Disorder | Inh | Gene | Protein (function) | Clinical features |
| Chédiak–Higashi syndrome | AR | LYST | See Table 60.5 | See Table 60.5 |
| Griscelli syndrome type 2 | AR | RAB27A | ||
| Familial hemophagocytic lymphohistiocytosis | AR | PRF1 | Perforin (major cytolytic granule protein of CTL and NK cells) |
|
| AR | UNC13D | Unc13 homolog D (primes cytolytic granules for secretion) | ||
| AR | STX11 | Syntaxin 11 (involved in cytolytic granule trafficking and fusion) | ||
| AR | STXBP2 | Syntaxin-binding protein 2 (regulates cytolytic granule trafficking) | ||
| X-linked lymphoproliferative syndrome (Duncan disease) | XR | SH2D1A | Signaling lymphocyte activation molecule-associated protein (adaptor protein that regulates B-, T- and NK-cell function; particularly important for immune responses to EBV) |
|
| XR | XIAP | X-linked inhibitor of apoptosis | ||
| EBV-associated lymphoproliferative syndrome | AR | ITK | IL-2-inducible T-cell kinase | |
| XMEN ( X -linked immunodeficiency with m agnesium defect, E BV infection, and n eoplasia) | XR | MAGT1 | Magnesium transporter 1 |
|
| STK4 deficiency | AR | STK4 | Serine/threonine kinase 4 |
|
| Autoimmune lymphoproliferative syndrome (Canale–Smith syndrome) and related conditions * | AD >AR | TNFRSF6 | CD95 (Fas; cell-surface apoptosis receptor) |
|
| AD >AR | TNFSF6 | CD95L (Fas ligand) | ||
| AR | FADD | Fas-associated death domain | ||
| AD > AR | CASP10 | Caspase 8 and 10 (proteases in intracellular apoptosis cascade) | ||
| AR | CASP8 | |||
| Mosaic | NRAS , KRAS | NRAS, KRAS (gain of function leads to decreased lymphocyte apoptosis) | ||
| AR | PRKCD | Protein kinase C δ (regulates B-cell proliferation) | ||
| AD | CTLA4 | Cytotoxic T lymphocyte-associated protein 4 (costimulation of T cells) | ||
| Immunodeficiency with lymphoid proliferation | AR | IL2RA | IL-2 receptor α-chain (apoptosis of developing T cells in the thymus) |
|
* LRBA mutations (see Table 60.15 ) and gain-of-function CARD11 or STAT3 mutations can also lead to autoimmune lymphoproliferative syndrome-like disorders.
Treatment
Hematopoietic stem cell transplantation (HSCT) can reverse the immunodeficiency of CHS and may also be performed following immunochemotherapy with etoposide, corticosteroids, and cyclosporine to abrogate the lymphoproliferative accelerated phase. However, HSCT has no effect on pigmentary alteration and does not halt neurologic degeneration. Management of the CHS is otherwise largely supportive, with administration of prophylactic antibiotics to prevent recurrent infections.
Complement Disorders
- ▪
Deficiency or dysfunction of early complement components increases susceptibility to pyogenic infections caused by encapsulated bacteria and autoimmune disorders, especially systemic lupus erythematosus
- ▪
Deficiency of late complement components leads to a markedly increased risk of neisserial infections
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


