Abstract
The porphyrias comprise a clinically and genetically heterogeneous group of diseases mostly arising from a genetically determined dysfunction of specific enzymes in the heme biosynthetic pathway. Based on the presence or absence of cutaneous symptoms and/or life-threatening acute neurological attacks, the different types of porphyria can be classified into either cutaneous and non-cutaneous forms or acute and non-acute forms. Establishing an accurate diagnosis can sometimes be difficult because the porphyrias often manifest with a broad, but nonspecific, spectrum of clinical symptoms that mimic several other disorders. Furthermore, biochemical assays of urine, feces, and blood can produce overlapping results. However, recent advances within the field of molecular genetics have helped to overcome these laboratory pitfalls. In diagnostically difficult cases, the correct diagnosis can be established via genetic analyses. Due to the various facets of the porphyrias, diagnosis and treatment often require interdisciplinary collaborations that include counseling of patients and their families.
Keywords
congenital erythropoietic porphyria, cutaneous porphyria, erythropoietic protoporphyria, heme, heme biosynthesis, hereditary coproporphyria, porphyria, porphyria cutanea tarda, variegate porphyria, X-linked dominant protoporphyria, pseudoporphyria, bullous dermatosis of dialysis
- ▪
The porphyrias result from dysfunction of enzymes involved in heme biosynthesis
- ▪
The different types can be classified into either acute versus non-acute or cutaneous versus non-cutaneous forms
- ▪
Cutaneous symptoms exclusively involve sun-exposed areas of the body
- ▪
Life-threatening neurologic attacks can occur in the acute porphyrias
- ▪
Diagnosis of specific types of porphyria may at times be difficult because clinical symptoms are often nonspecific and biochemical findings can overlap
- ▪
The genetic bases of the porphyrias have been well characterized, facilitating accurate molecular diagnosis and genetic counseling
Introduction
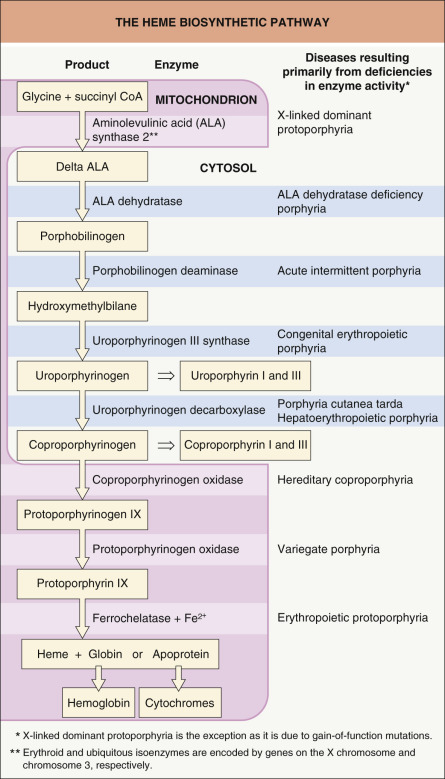
The porphyrias are metabolic disorders, each arising from a predominantly hereditary deficiency of one of the eight enzymes of the porphyrin-heme biosynthetic pathway ( Fig. 49.1 ) . Traditionally, these disorders are subdivided into erythropoietic and hepatic forms, according to the major site of expression of the enzyme deficiency. From a dermatologist’s perspective, the porphyrias can be classified into cutaneous and non-cutaneous forms ( Table 49.1 ), whereas from a general clinician’s point of view, it is more appropriate to classify the porphyrias into acute and non-acute forms, thereby emphasizing the presence or absence of potentially life-threatening acute neurologic attacks ( Table 49.2 ) . Therefore, we will adhere to the latter classification throughout this chapter.
| CLASSIFICATION OF THE PORPHYRIAS INTO CUTANEOUS AND NON-CUTANEOUS FORMS | |
|---|---|
| Cutaneous | Non-cutaneous |
|
|
| CLASSIFICATION OF THE PORPHYRIAS INTO ACUTE AND NON-ACUTE FORMS | |||
|---|---|---|---|
| Disorder | Incidence | Age of onset | Important aspects |
| Acute porphyrias | |||
| Acute intermittent porphyria | 0.5–1 per 100 000 | Second to fourth decade of life; rarely before puberty | Most common acute porphyria worldwide; acute neurologic attacks but no photosensitivity/cutaneous manifestations |
| Variegate porphyria | ~1 per 300 in South Africa; relatively rare elsewhere | Second to third decade of life; usually not before puberty | Cutaneous manifestations similar to porphyria cutanea tarda , and acute attacks similar to acute intermittent porphyria can occur (neurocutaneous porphyria); founder mutations identified in South Africa and Chile |
| Hereditary coproporphyria | Very rare (<75 cases reported) | Usually not before puberty | Acute attacks similar to acute intermittent porphyria; cutaneous manifestations, including erythema and blistering in sun-exposed areas, can occur (neurocutaneous porphyria) |
| ALA-D deficiency porphyria | Extremely rare (<10 cases reported) | Early and late onset have been described | Neurologic symptoms similar to acute intermittent porphyria can occur; no photosensitivity/cutaneous manifestations |
| Non-acute porphyrias (all can have cutaneous features) | |||
| Porphyria cutanea tarda | Most common porphyria worldwide | Third to fourth decade of life; usually not before puberty | Acquired and hereditary variants exist; moderate to severe photosensitivity; cutaneous findings in sun-exposed areas include skin fragility, erosions, crusts, vesicles and bullae, milia, scarring, hyperpigmentation and hypertrichosis; indistinguishable from variegate porphyria |
| Erythropoietic protoporphyria | Second-highest incidence of the cutaneous porphyrias | Early childhood (1 to 4 years); late onset extremely rare | Cutaneous manifestations include erythema, edema, crusts, purpura, skin thickening, and waxy scars, primarily on the dorsal hands and face; usually no blistering; in ~5% of patients, severe liver disease can occur; cholelithiasis; palmar keratoderma in rare AR form; adult-onset form associated with underlying MPD or MDS |
| Congenital erythropoietic porphyria | Very rare (~170 cases reported) | Infancy/first decade of life | Very severe clinical course; in sun-exposed sites, vesicles and bullae, erosions, ulcerations, crusts, milia, scarring, hyperpigmentation and hypertrichosis; mutilation; hemolytic anemia; hepatosplenomegaly; porphyrin deposition in bones and teeth (erythrodontia); pink, red or violet staining of diapers |
| Hepatoerythropoietic porphyria | Extremely rare (~35 cases reported) | Early infancy | Recessive variant of porphyria cutanea tarda; markedly increased photosensitivity and severe clinical course possible; in sun-exposed sites, vesicles and bullae, skin fragility, erosions, crusts, milia, scarring and hypertrichosis; mutilation can occur; diapers stained by darkly colored urine |
| X-linked dominant protoporphyria | Very rare | Early childhood | Skin symptoms indistinguishable from erythropoietic protoporphyria; most recent type of porphyria to be recognized; liver disease appears to occur more frequently than in erythropoietic protoporphyria |
The porphyrias are of particular dermatologic interest because most forms have characteristic cutaneous manifestations that often allow a presumptive diagnosis. Laboratory tests are then used to confirm the suspected diagnosis. The genetic defects underlying the porphyrias have been well characterized ( Table 49.3 ), facilitating molecular diagnosis and genetic counseling for affected families.
| GENETIC ASPECTS OF THE PORPHYRIAS | |||
|---|---|---|---|
| Disorder | Gene locus | Protein product and gene symbol | Mode of inheritance |
| Acute porphyrias | |||
| Acute intermittent porphyria | 11q23.3 | Porphobilinogen deaminase; PBGD | AD |
| Variegate porphyria | 1q22–23 | Protoporphyrinogen oxidase; PPOX | AD |
| Hereditary coproporphyria | 3q12 | Coproporphyrinogen oxidase; CPO | AD |
| ALA-D deficiency porphyria | 9q34 | δ-Aminolevulinic acid dehydratase; ALAD | AR |
| Non-acute porphyrias | |||
| Porphyria cutanea tarda | 1p34 | Uroporphyrinogen decarboxylase; UROD | AD (up to 25% of cases); otherwise acquired |
| Erythropoietic protoporphyria | 18q21.3 | Ferrochelatase; FECH | AD * ; rarely, AR |
| Congenital erythropoietic porphyria | 10q25.2–q26.3 | Uroporphyrinogen III synthase; UROS | AR |
| Hepatoerythropoietic porphyria | 1p34 | Uroporphyrinogen decarboxylase; UROD | AR |
| X-linked dominant protoporphyria | Xp11.21 | δ-Aminolevulinic acid synthase 2; ALAS2 (gain-of-function mutations) | XLD |
* Actually semi-dominant inheritance with a mutation in one allele and a specific intronic polymorphism in the second allele (“hypomorphic” allele; see text).
History
In 1874, Schultz described a man with a history of cutaneous photosensitivity, accompanied by the excretion of wine-red urine . Subsequently, Baumstark detected urinary pigments, which he named “urorubrohaematin” and “urofuscohaematin” . Günther established the first classification of the porphyrias in 1911, recognizing them as hereditary metabolic disorders characterized by increased porphyrin excretion. He distinguished between two different forms: (1) haematoporphyria acuta, characterized by acute neurovisceral attacks without skin lesions; and (2) haematoporphyria congenita and chronica, both disorders having cutaneous findings in sun-exposed areas of the body . In 1937, the terms “acute intermittent porphyria” and “porphyria cutanea tarda” were introduced. Over the next four decades, variegate porphyria (in 1953), hereditary coproporphyria (in 1955), erythropoietic protoporphyria (in 1961), hepatoerythropoietic porphyria (in 1969), and δ-aminolevulinic acid (ALA) dehydratase deficiency porphyria (in 1979) were described . Most recently, X-linked dominant protoporphyria was recognized .
Epidemiology
The porphyrias are rare disorders, occurring in all races and both sexes. Whereas some forms appear during infancy, other variants usually do not present until puberty or adulthood. Prevalence rates vary from 0.1 to 10 per 100 000 individuals (see Table 49.2 ) . However, the precise prevalence of the various porphyrias is not known, due primarily to geographic differences, the likelihood of underdiagnosis, and the incomplete penetrance of the dominantly inherited porphyrias.
Pathogenesis
Mutations in any of the genes encoding the enzymes in the heme biosynthetic pathway can lead to a pathologic accumulation and excess excretion of porphyrins and/or porphyrin precursors, as a result of enzyme dysfunction . Except for patients with acquired porphyria cutanea tarda, all forms of porphyria are inherited as monogenetic traits (see Table 49.3 ).
Cutaneous Findings
To date, no single factor can explain the various photosensitivity patterns evoked by porphyrins plus visible and UV radiation. Nonetheless, a number of cellular and soluble factors are thought to be involved, including reactive oxygen species, certain cell types (e.g. erythrocytes, mast cells, polymorphonuclear cells, fibroblasts), and soluble mediators (e.g. components of the complement system or factor XII-dependent pathways, eicosanoids), as well as matrix metalloproteinases . Most likely, interactions between these factors contribute to the development of cutaneous lesions.
Within the Soret band (400 to 410 nm), porphyrins absorb light energy most efficiently and enter into an excited molecular state. There is also less prominent absorption of adjacent UV wavelengths and other minor visible bands. These light-excited porphyrins can then in turn transfer this absorbed energy to oxygen molecules thereby generating highly reactive oxygen species. Cellular and tissue damage induced by photoactivated porphyrins is believed to result primarily from the formation of reactive singlet oxygen and free radicals, with subsequent lipid peroxidation and protein cross-linking .
The type of cellular damage depends on the solubility and tissue distribution of the porphyrins. Accumulation of water-soluble uro- and coproporphyrins leads to blistering, as is seen in most of the cutaneous porphyrias (e.g. porphyria cutanea tarda, variegate porphyria). In contrast, accumulation of lipophilic protoporphyrins leads to an immediate cutaneous burning sensation after exposure to the appropriate wavelengths of light, accompanied by erythema and edema, as seen in erythropoietic protoporphyria .
Acute Porphyric Attack
Two porphyria variants, ALA dehydratase deficiency porphyria and acute intermittent porphyria, are not associated with cutaneous findings. The dysfunctional enzymes in these two porphyrias are involved in early steps of heme biosynthesis, and their substrates, ALA and porphobilinogen (PBG), respectively, are non-phototoxic porphyrin precursors (see Fig. 49.1 ). However, both acute intermittent porphyria and ALA dehydratase deficiency porphyria can manifest with life-threatening acute neurologic attacks (see Table 49.2 ). It appears that ALA and PBG, which are excreted in massive amounts from the liver during an acute attack, are extremely neurotoxic. Lacking appropriate barrier protection, the autonomic and peripheral nervous systems are particularly susceptible to the toxic effects .
Of note, porphyrin abnormalities are also observed in the setting of lead poisoning, sideroblastic and hemolytic anemia, iron deficiency, renal failure, cholestasis, liver disease, and gastrointestinal hemorrhage. However, associated photosensitivity has only been documented in rare cases of sideroblastic anemia .
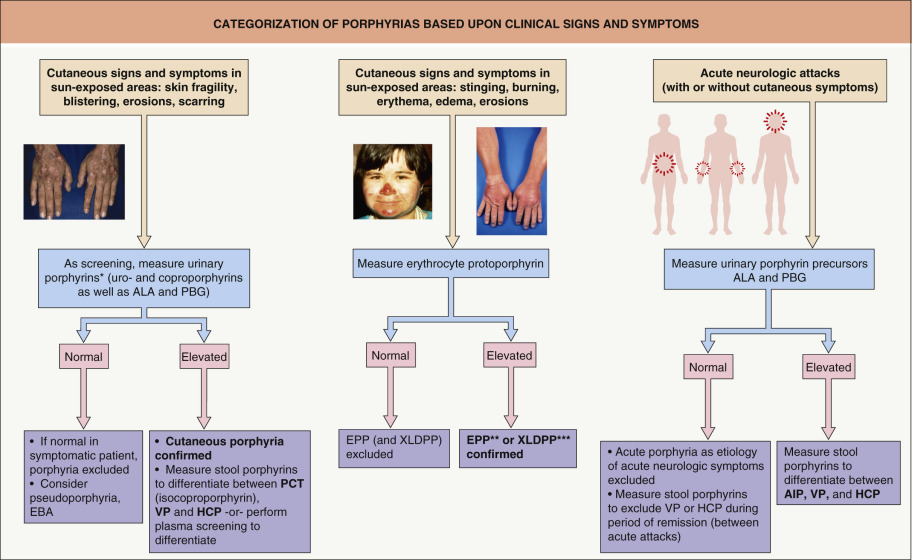
Clinical and Laboratory Investigation
In establishing the diagnosis of a specific type of porphyria, the medical evaluation involves up to four steps:
- •
a thorough clinical assessment that includes a family history and physical examination, with particular reference to sun-exposed sites
- •
biochemical measurement of porphyrins and porphyrin precursors in urine, feces, blood, and/or plasma ( Table 49.4 )
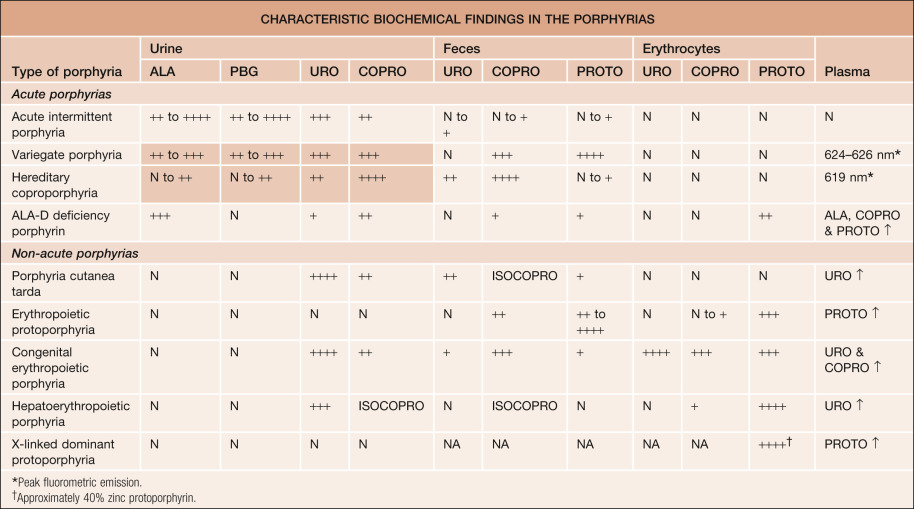
Table 49.4
Characteristic biochemical findings in the porphyrias.
In patients with variegate porphyria or hereditary coproporphyria who have no symptoms (cutaneous or systemic), urine porphyrins may not be elevated (shaded areas). ALA, aminolevulinic acid; ALA-D, aminolevulinic acid dehydratase; COPRO, coproporphyrin; ISOCOPRO, isocoproporphyrin; PBG, porphobilinogen; PROTO, protoporphyrin; URO, uroporphyrin. N = normal; NA, not available; + = above normal range; ++ = slightly elevated; +++ = highly elevated; ++++ = very highly elevated; ↑ = increase.
- •
determination of the activity of specific enzymes (specialized laboratories)
- •
DNA mutational analysis (specialized laboratories).
With regard to biochemical analyses, markedly elevated urinary levels of the porphyrin precursors ALA and PBG can be found during an acute attack. However, asymptomatic “carriers” (who harbor mutations) are rarely detected via measurement of urinary porphyrin precursors because the latter can exhibit significant variability and levels may be only slightly elevated or even normal during the intervals between acute attacks. In addition, diagnoses based upon measurements of enzymatic activity in fibroblasts or lymphocytes are not always conclusive. This is because an overlap exists between the values obtained in patients, asymptomatic carriers, and normal control individuals .
Therefore, the ability to detect specific genetic mutations has made a significant impact on the precise diagnosis of porphyrias and the ability to provide accurate genetic counseling .
The Non-Acute Porphyrias
The non-acute porphyrias consist of porphyria cutanea tarda (PCT), erythropoietic protoporphyria (EPP), X-linked dominant protoporphyria (XLDPP), congenital erythropoietic porphyria (CEP), and hepatoerythropoietic porphyria (HEP). All these porphyrias present primarily with cutaneous findings (see Tables 49.1 & 49.2 ) . While histologic examination of involved skin is not required to confirm a presumptive diagnosis of any of the cutaneous porphyrias, a biopsy specimen may have been obtained to exclude other entities in the differential diagnosis. Occasionally, the pathologist may identify histologic features that support the diagnosis of cutaneous porphyria even though the clinician suspected another disorder, e.g. epidermolysis bullosa acquisita.
Porphyria Cutanea Tarda (PCT)
Worldwide, PCT is the most common type of porphyria. It results from decreased activity of uroporphyrinogen decarboxylase, the fifth enzyme in heme biosynthesis (see Fig. 49.1 ) .
At least two types of PCT can be distinguished: (1) a sporadic (acquired) variant, designated type I PCT, in which the dysfunctional enzyme is exclusively expressed in the liver; and (2) an autosomal dominant familial (hereditary) variant, designated type II PCT, in which the catalytic enzymatic defect is detected in all tissues (see Table 49.3 ). In most countries, the ratio of type I to type II PCT is currently estimated to be approximately 3 : 1 to 4 : 1 . Interestingly, Elder reported several families with typical clinical and biochemical features of overt PCT but with normal uroporphyrinogen decarboxylase activity in their red blood cells. This variant of the disease has been designated type III PCT. Indeed, there is increasing evidence that some facets of the etiology of PCT have yet to be completely elucidated.
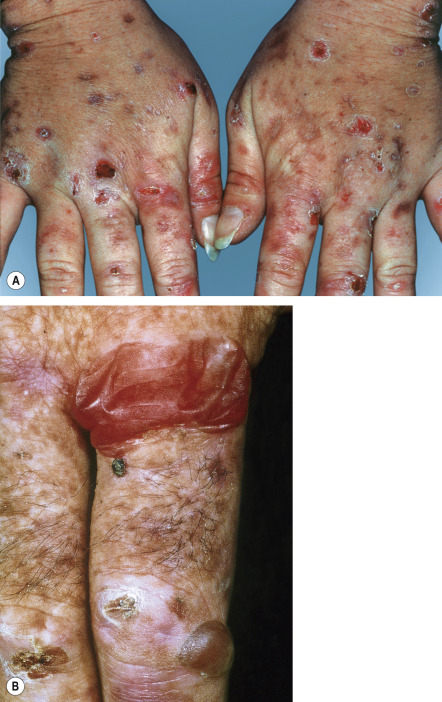
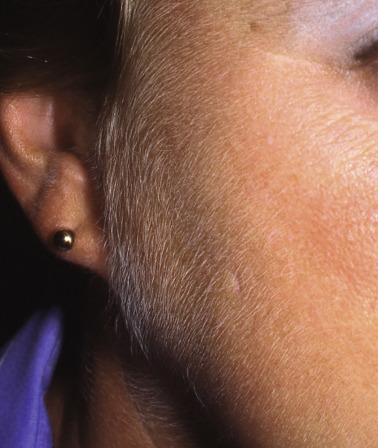
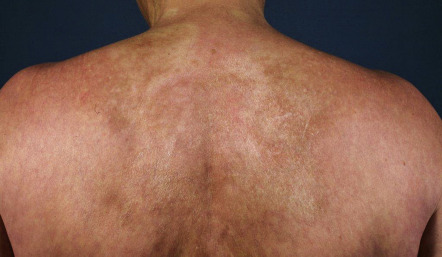
The cutaneous manifestations of PCT include increased photosensitivity and skin fragility as well as blistering, erosions, crusts, milia, and scars in sun-exposed sites ( Fig. 49.3 ; see Table 49.2 ). Additionally, postinflammatory hyperpigmentation, hypertrichosis ( Fig. 49.4 ), scarring alopecia, and morpheaform and sclerodermoid changes ( Fig. 49.5 ) can be observed.