Abstract
Pathologic reactions of the skin to light can arise from multiple etiologies including: immunologic photodermatoses, e.g. polymorphous light eruption, solar urticaria, actinic prurigo, hydroa vacciniforme, and chronic actinic dermatitis; photosensitivity due to defective DNA repair, e.g. xeroderma pigmentosum, trichothiodystrophy, Bloom syndrome, Rothmund–Thomson syndrome; dermatologic disorders that may be aggravated by sun exposure; and phototoxic and photoallergic reactions due to exogenous and endogenous photosensitizers, especially drugs. Normal responses to sunlight exposure include sunburn, immediate pigment darkening, tanning, vitamin D synthesis, immunosuppression, and chronic changes such as wrinkling, solar elastosis, and photocarcinogenesis.
Keywords
photodermatoses, polymorphous light eruption, polymorphic light eruption, solar urticaria, actinic prurigo, hydroa vacciniforme, chronic actinic dermatitis, ultraviolet radiation, defective DNA repair, photoaggravated dermatoses, phototoxicity, photoallergy, drug-induced photosensitivity, photoallergic drug reaction, phototoxic drug reaction, allergic photocontact dermatitis, photopatch testing, xeroderma pigmentosum, Cockayne syndrome, trichothiodystrophy, Bloom syndrome
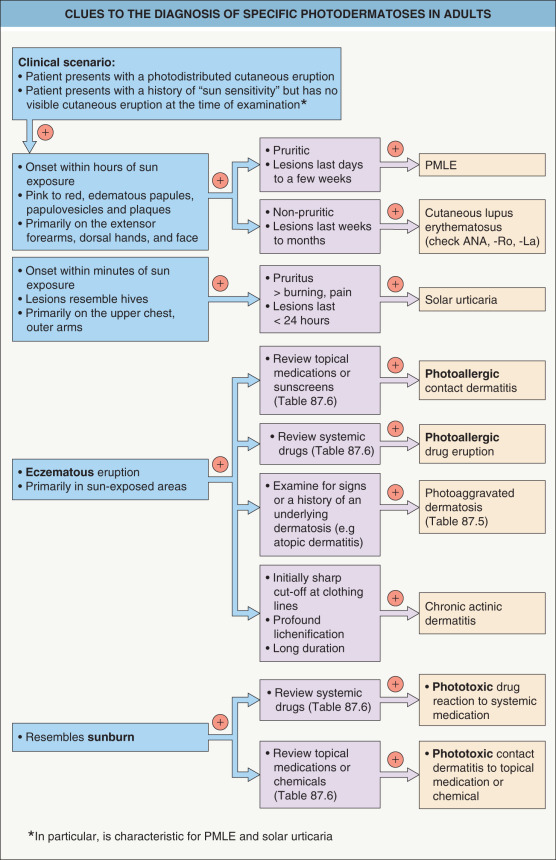
All skin can react to light because it contains molecules (chromophores) whose chemical structures are capable of absorbing ultraviolet radiation (UVR) or other electromagnetic energy. This absorbed energy is then either re-emitted harmlessly as biologically inactive radiation or diverted to driving thermochemical reactions, leading to molecular, cellular, tissue, and clinical changes. These alterations are subsequently repaired or result in permanent effects. Nucleic acids are the most ubiquitous chromophores, initiating UVR-induced changes including sunburning, tanning, hyperplasia, aging, and carcinogenesis. Photodermatoses result from either abnormal tissue responses following absorption by endogenous molecules or expected responses to absorption by porphyrins or photosensitizing drugs or chemicals ( Table 87.1 ). Clues to the diagnosis of specific photodermatoses are provided in Fig. 87.1 .
| CLASSIFICATION OF PHOTODERMATOSES |
|
Abnormal Cutaneous Effects to Light
Polymorphous light eruption is consistently the most common photodermatosis, based upon relative frequency analyses from photodermatology centers worldwide; it is followed (in decreasing order of frequency) by photoaggravated dermatoses, drug-induced photosensitivity, chronic actinic dermatitis, and solar urticaria . It has been well documented that photodermatoses have a significant negative effect on patients’ quality of life .
Immunologically-Mediated Photodermatoses
This group of disorders is outlined in Table 87.2 .
| IDIOPATHIC, POSSIBLY IMMUNOLOGICALLY MEDIATED PHOTODERMATOSES |
|
Polymorphous light eruption
▪ Polymorphic light eruption ▪ Benign summer light eruption (clinical variant) ▪ Juvenile spring eruption (clinical variant)
- ▪
The most common photodermatosis
- ▪
Papules, vesicles or plaques within hours of sun exposure; lasts for a few days
- ▪
Action spectra: UVB, UVA, and rarely visible light
- ▪
Management: photoprotection and narrowband (NB)-UVB; occasionally, brief courses of topical or oral corticosteroids for acute attacks
Introduction and history
Polymorphous light eruption (PMLE) is a common, sunlight-induced eruption affecting individuals of all races and skin colors . Attacks are intermittent and follow minutes to hours (rarely days) of exposure of the skin to sunlight or artificial UVR. Non-scarring, pruritic, erythematous papules, papulovesicles, vesicles, or plaques then develop hours later (occasionally within minutes). The eruption is generally most severe in the spring and early summer, especially in temperate climates. It improves by autumn and then usually completely disappears over the winter. In some patients, PMLE may only occur when they travel to tropical regions.
PMLE was first described by Carl Rasch in 1900 and mentioned again by Haxthausen in 1918.
Epidemiology
The prevalence of PMLE in the general population is inversely related to latitude, being highest in Scandinavia (22%), high in the UK and northern US (10–15%), and low in Australia (5%) and equatorial Singapore (~1%) . This is probably due to the development of UVR-induced immunologic tolerance, commonly referred to as “hardening”, secondary to constant year-round solar exposure in sunny climates. Women are affected slightly more often than men, with the second and third decades being the most common times of onset.
Pathogenesis
Attacks of PMLE are triggered by exposure to UVR (and perhaps, on occasion, visible light) from sunlight or other sources such as tanning beds. Spring and summer sunlight in temperate regions is most likely to induce outbreaks, but generally at amounts lower than the minimal erythema dose (MED). The action spectrum ranges from UVB to UVA and rarely to visible light. Photoprovocation studies, requiring repeated exposures of UVR to the same sites over several days, have shown a positive response in 50% of patients to NB-UVB, 50% to UVA, and 80% to both UVB and UVA . However, patients with PMLE typically have normal MEDs to UVB, UVA, and visible light .
PMLE appears to represent a delayed-type hypersensitivity (DTH) response to as-yet-undefined, endogenous cutaneous, photoinduced antigens . Timed biopsy specimens, following solar-simulated irradiation (~0.6 MED), have shown perivascular infiltrates of lymphocytes, primarily CD4 + (within hours) and CD8 + (within days), in association with an increased number of dermal and epidermal antigen-presenting cells . Following UV exposure, reductions in IL-4 and IL-10 production as well as in infiltrates of neutrophils and mast cells have been observed in PMLE .
Susceptibility to PMLE appears to be genetic, with up to 70% of the population having a propensity for developing this condition, but not all expressing it, because of variations in disease penetrance . There appears to be a resistance to the normal UVR-induced suppression of the induction , but not the elicitation , of cutaneous DTH responses. As a result, in PMLE, a lesser degree of cutaneous immunosuppression following UVR exposure (compared to normal individuals) likely results in a persistent ability to mount a DTH response against UVR-altered endogenous cutaneous molecules, leading to the development of clinical lesions. Possible underlying mechanisms include: (1) an under-expression of apoptotic-cell clearance genes in keratinocytes, thus prolonging antigen presence; and/or (2) inefficient free radical removal, thereby increasing photoantigen production. In evolutionary terms, UVR-induced cutaneous immunosuppression may protect against developing disruptive PMLE, but it can increase the risk of skin cancer; of note, PMLE patients appear to have a reduced tendency to develop such cancers .
Clinical features
PMLE occurs most commonly during the spring and early summer, following minutes to hours (occasionally days) of sun exposure . Outbreaks may also occur after exposure to snow-reflected sunlight, tanning beds, or UV phototherapy. The eruption develops hours (occasionally minutes) after UVR exposure; it then fades over one to several days or occasionally weeks if the exposure is ongoing. However, the likelihood of occurrence often diminishes or ceases over the summer or a lengthy sunny vacation, presumably due to the development of immunologic tolerance, sometimes called “hardening” .
Typically, some, but virtually never all, of the exposed skin is affected. Lesions are usually symmetrically distributed in a patchy fashion. Areas that are normally continuously exposed to sunlight (e.g. face) are often spared, although not always ( Fig. 87.2 ). The distribution pattern and type of lesions in a given patient are usually consistent over time. Areas commonly affected are the neck, outer aspects of the arms, and dorsal hands ( Fig. 87.3 ), but there may be more widespread involvement of sun-exposed sites ( Fig. 87.4 ).
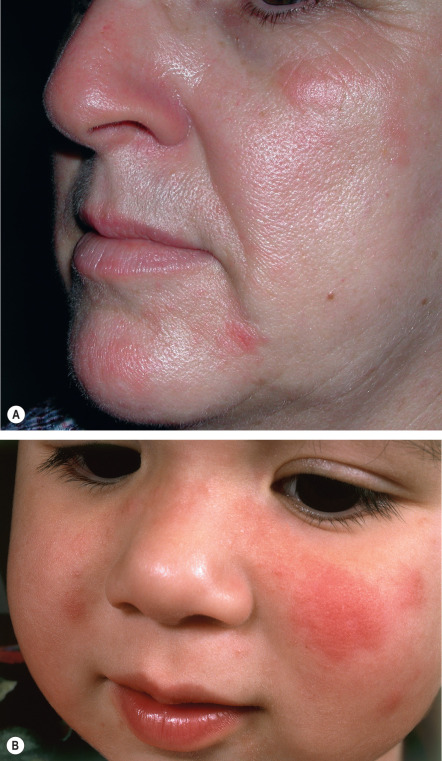
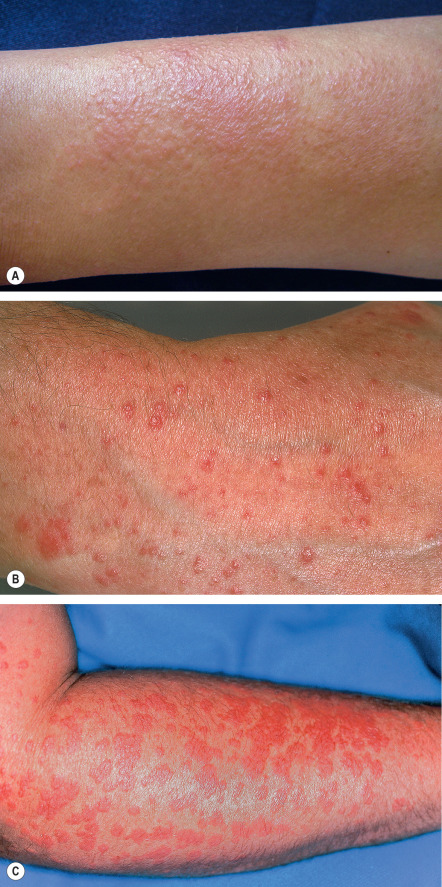
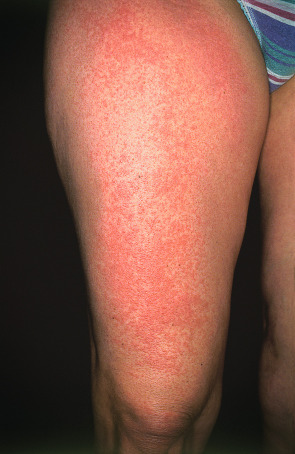
Lesions are polymorphous and vary widely amongst affected individuals. Clinically, mildly to markedly pruritic, grouped, erythematous or skin-colored papules of varying sizes, sometimes coalescing into large, smooth or unevenly surfaced plaques, are seen. In darkly pigmented individuals, the most common morphology is grouped, pinhead-sized papules in sun-exposed areas . Vesicles, bullae, papulovesicles, and confluent edematous swelling are additional manifestations; rarely, only pruritus occurs. Occasionally, particularly in boys, the helices of the ears may be principally affected in a form of PMLE referred to as juvenile spring eruption. Vesicles are commonly observed in this variant and rarely patients develop fever, general malaise, headache, and nausea. PMLE may be lifelong; however, in a 32-year follow-up of 94 patients, the disease improved or resolved in 58% over a period of 16 years, and in 75% over a period of 32 years.
Pathology
There is variable epidermal spongiosis and a superficial and deep, perivascular and periappendageal, lymphohistiocytic dermal infiltrate, often with scattered eosinophils and neutrophils. Significant papillary dermal edema occurs commonly ( Fig. 87.5 ). Rarely, difficulty in distinction from cutaneous lupus may occur if interface changes are significant, or from lymphoma if the dermal inflammatory cell infiltrate is marked.
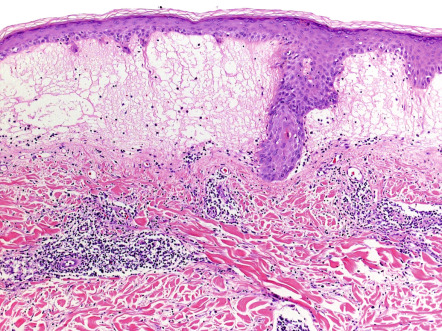
Differential diagnosis
PMLE can often be distinguished from lupus erythematosus (LE), photoaggravated dermatoses (e.g. atopic dermatitis, seborrheic dermatitis), solar urticaria, and rarely, erythropoietic protoporphyria (EPP) by the natural history and clinical appearance of the cutaneous lesions. Occasionally, clinicopathologic correlation and laboratory evaluation are required ( Table 87.3 ; see Fig. 87.1 ). For photoaggravated dermatoses, the characteristic clinical features of the primary disorder usually allow for distinction.
| EVALUATION OF PHOTOSENSITIVITY – POSSIBLE LABORATORY INVESTIGATIONS |
|
Treatment
PMLE in its milder forms may respond to photoprotection, including the use of broad-spectrum, high SPF sunscreens and physical barriers. In patients with more severe disease, hardening via prophylactic, two-to-three times weekly sessions of NB-UVB, usually for 15 sessions in the spring, may be effective for several months . An initial starting dose of 50% of the MED for NB-UVB is recommended, with the dose then increased by 10–15% per treatment. Oral prednisone (0.5–1 mg/kg) may be used during the initial seven to ten days of phototherapy to minimize photoexacerbation. Once hardening is achieved, it may be possible to maintain the effect by having patients expose themselves weekly to sunlight between 10 AM and 2 PM for 15–20 minutes (without sunscreen) for the remainder of the sunny season. However, some patients find that they do not need to intentionally seek sun exposure to maintain the hardening.
Other therapeutic options for prevention of PMLE include oral prednisone (<0.5 mg/kg, usually given for 5–7 days during vacations to a sunny climate) and hydroxychloroquine . For symptomatic eruptions, topical corticosteroids and oral prednisone can be prescribed.
Actinic prurigo
▪ Hutchinson’s summer prurigo ▪ Familial (or hereditary) polymorphous light eruption of American Indians ▪ Hydroa aestivale
- ▪
Severe form common in Native Americans, often with cheilitis and conjunctivitis
- ▪
Can occur in all races
- ▪
Childhood onset; often resolution by adolescence, but can persist indefinitely
- ▪
Intensely pruritic, crusted papules or nodules in sun-exposed sites
- ▪
Strong association with HLA-DR4 (DRB1*0401) and subtype DRB1*0407
- ▪
Management: photoprotection, NB-UVB, topical calcineurin inhibitors, and thalidomide
Introduction and history
Actinic prurigo is a fairly uncommon, sunlight-induced, pruritic and crusted, papular or nodular eruption involving uncovered and, to a lesser extent, covered skin . It was first described in the 1960s and 1970s, and was noted to affect North, Central and South Americans of indigenous or mixed race.
Hutchinson first described this disorder in 1879, followed in 1918 by Haxthausen, who described it as a PMLE variant. In 1968, Londoño coined the term “actinic prurigo”.
Epidemiology
Actinic prurigo is particularly common in Native Americans, especially Mestizos (mixed American Indian and European ancestry) living at high altitudes. It also occurs, but much less commonly, in Europe and Asia. A familial form is relatively common.
Pathogenesis
Actinic prurigo is triggered by UVR exposure and therefore typically flares during the spring and summer, especially in temperate climates. Phototesting results are abnormal in approximately two-thirds of patients and cutaneous lesions can be provoked by UVB or UVA.
The immunologic nature of actinic prurigo is supported by a study of Mexican Mestizo patients, where the remarkable therapeutic efficacy of thalidomide has been shown to be associated with an inhibition of TNF-α synthesis and the modulation of interferon-γ-producing CD3 + cells . The presence of a dense lymphocytic infiltrate and lymphoid follicles in lesional biopsy specimens from the lips provides further support for this concept .
Actinic prurigo may represent a persistent variant of PMLE, as clinical conversion between actinic prurigo and PMLE has been observed, and in family studies, the two disorders are closely linked . There is also a strong association of actinic prurigo with HLA-DR4 (DRB1*0401) and subtype DRB1*0407 , and this may be the genetic factor that transforms PMLE into actinic prurigo. Indeed, the presence of this HLA subtype is strongly suggestive of the diagnosis.
Clinical features
Actinic prurigo usually appears during childhood and is more frequently seen in girls. It often resolves by adolescence, but may persist indefinitely . The eruption is usually worse during the summer in temperate climates, but may not always be clearly related to sun exposure. In tropical regions of Latin America, it can persist year-round.
Erythematous papules or nodules, sometimes with hemorrhagic crusts due to excoriation, are present in sun-exposed sites, especially the face (including the nose) and the distal limbs ( Fig. 87.6 ). Lesions may be scattered or densely distributed, and there are sometimes eczematous features or lichenification. Fine linear or pitted scars may appear as facial papules heal. In patients from the UK, involvement of covered sites, particularly the buttocks, has been well described ( Fig. 87.7 ). Cheilitis and conjunctivitis are common, with cheilitis often being the only clinical manifestation ( Fig. 87.8 ) .
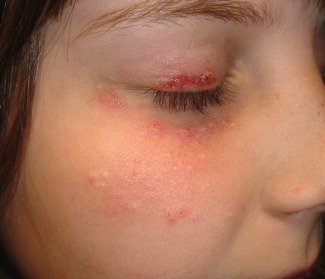
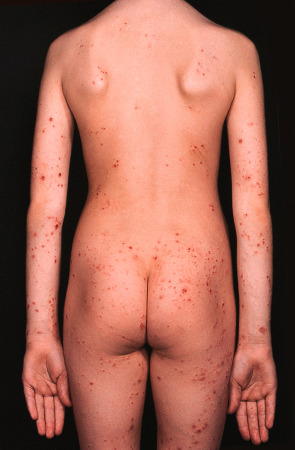
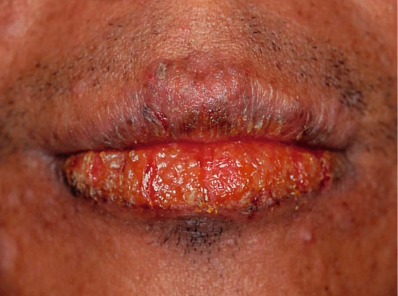
Pathology
In early lesions, there is epidermal spongiosis, acanthosis, and a dermal perivascular mononuclear cell infiltrate with occasional eosinophils, but, unlike in PMLE, papillary edema is usually absent . Later, crusts, more pronounced acanthosis, variable lichenification, excoriations, focal dermal papillary fibrosis, and a heavier mononuclear cell infiltrate can be seen. These latter features are nonspecific and may resemble those of chronic dermatitis, prurigo nodularis, or, very rarely, cutaneous B-cell lymphoma. Dermal lymphoid follicles have been observed in biopsy specimens taken from the lips . Direct immunofluorescence microscopy is negative.
Differential diagnosis
The history and clinical features often suggest the diagnosis. Clinicopathologic correlation and laboratory evaluation aid in excluding LE and childhood porphyrias (see Table 87.3 ). Prurigo nodularis is not accentuated in sun-exposed skin.
Treatment
Rigorous photoprotection is helpful. For milder disease, topical corticosteroids and topical tacrolimus may be used. NB-UVB should be considered next, with protocols similar to those for PMLE (see above), although access to phototherapy may be limited. Resistant disease is best treated with oral thalidomide (50–100 mg nightly) , administered for several weeks until remission is achieved and then tapered to as low a maintenance dose as possible (e.g. 50 mg every second or third night). However, the risks of teratogenicity and peripheral neuropathy require very careful patient selection and supervision (see Ch. 130 ). Other possible systemic therapies include corticosteroids, azathioprine, and cyclosporine. The role of TNF-α inhibitors is still being investigated.
Hydroa vacciniforme
▪ Bazin’s hydroa vacciniforme
- ▪
Very rare, childhood-onset photodermatosis
- ▪
Papules and plaques develop umbilicated vesiculation, followed by hemorrhagic crusting (often severe) and varioliform scarring
- ▪
Epstein–Barr viral infection has been detected in a number of patients
- ▪
Management requires very careful photoprotection and perhaps low-dose phototherapy
Introduction and history
Hydroa vacciniforme (HV) is a rare, sunlight-induced, childhood-onset, intermittent but permanently scarring disorder. The condition was first reported by Bazin in 1862.
Epidemiology
HV is well recognized globally, including in the US, UK, Europe, Korea and Japan, and it has a predilection for lightly pigmented individuals. Its rarity and lack of specific diagnostic tests (apart from its characteristic early histology) have made precise assessment of its distribution difficult. The condition typically has its onset during childhood, affecting boys slightly more often than girls and with more severe disease in boys. The disorder can resolve during adolescence or early adulthood; rare familial cases have been noted .
Pathogenesis
Although tropical sunlight or summer sunlight in temperate regions is generally required to provoke the eruption, abnormal erythematous, or rarely vesicular, phototest responses to repeated monochromatic or broad-spectrum UVR exposure can be induced. The exact nature of the cutaneous reaction in HV is unknown. There have been multiple reports documenting Epstein–Barr virus (EBV) within cutaneous lesions of apparent HV . In these patients with chronic EBV infection, the clinical presentations ranged from “typical” HV to a severe HV-like eruption. While no hematologic abnormalities were detected in the former group, those in the latter group had NK-cell lymphocytosis, exaggerated reactions to mosquito bites, and hemophagocytic syndrome. In addition, several of the patients with severe HV-like eruptions have died of EBV-associated NK- or T-cell lymphoma or the hemophagocytic syndrome .
In a study from Japan, T cells positive for EBV RNA were detected in lesional skin infiltrates from 94% of children with clinically typical HV (17/18) and 11/11 patients with severe HV-like eruptions associated with systemic symptoms, but 0/32 skin biopsy samples from patients with various other dermatoses, including photodermatoses . While the relationship between EBV infection and true photosensitive HV has yet to be firmly established, it is likely that HV and severe HV-like eruptions represent a clinical spectrum.
Clinical features
Symmetrical, clustered, pruritic or stinging, erythematous macules develop in a photodistribution, especially on the face and dorsal aspects of the hands, within hours of summer sun exposure. Over the next several hours, the macules become tender papules or plaques surmounted by vesicles or bullae (often hemorrhagic), before umbilicating and condensing into hemorrhagic crusts over several days ( Fig. 87.9A,B ). Healing then occurs over a period of weeks, leaving individual or confluent, sometimes telangiectatic, varioliform scars ( Fig. 87.9C ). Generalized malaise with fever or headache rarely accompanies the attacks. However, quality of life in affected children is negatively impacted by their inability to play outdoors .
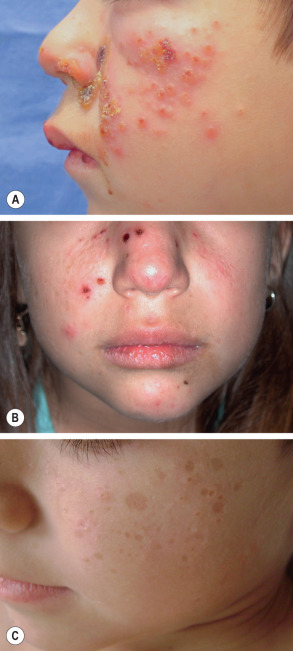
Pathology
Progressive epidermal spongiosis precedes findings that are almost pathognomonic: prominent, reticular keratinocyte degeneration; the formation of intraepidermal vesicles containing fibrin and acute inflammatory cells; and confluent epidermal and occasionally focal upper dermal necrosis ( Fig. 87.10 ) . Early on, a variably dense perivascular lymphohistiocytic infiltrate admixed with neutrophils is also seen, followed by neutrophilic invasion. Direct immunofluorescence microscopy findings are nonspecific. In situ hybridization for EBV RNA shows positivity within the lymphoid infiltrate.
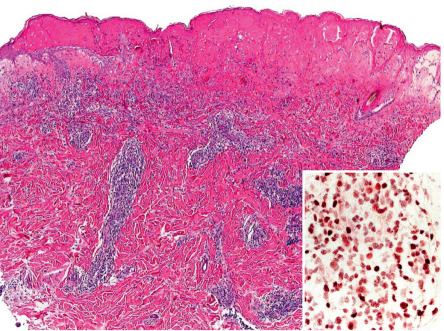
Differential diagnosis
While LE and childhood porphyrias can be excluded by clinicopathologic correlation and laboratory evaluation (see Table 87.3 ), herpes simplex is excluded by a negative PCR, DFA assay, or viral culture. In patients with severe HV-like eruptions associated with systemic EBV-related disease ( Fig. 87.11 ), the skin lesions typically become more severe and more widespread to include sun-protected sites; additional features include facial swelling, ulcerated nodular skin lesions, exaggerated reactions to mosquito bites, high-grade fevers and hepatosplenomegaly, as well as leukopenia, thrombocytopenia, elevated hepatic transaminases, and increased NK lymphocytes. A high circulating EBV DNA load is generally detected by PCR.
Treatment
HV is almost always refractory to treatment . Photoprotection, including the use of tinted windows, can be useful in mild to moderate disease. Anecdotally, the following have been claimed as helpful: broadband (BB)-UVB, NB-UVB, PUVA, β-carotene, antimalarials, azathioprine, thalidomide, cyclosporine, and dietary fish oil.
Chronic actinic dermatitis
▪ Photosensitivity dermatitis and actinic reticuloid (PD/AR) syndrome ▪ Persistent light reaction ▪ Actinic reticuloid (severe clinical variant) ▪ Photosensitivity dermatitis (milder UVB- and UVA-sensitive variant) ▪ Photosensitive eczema (milder UVB only-sensitive variant)
- ▪
In sun-exposed areas, chronic eczematous eruption (acute, subacute or chronic lichenified) or pseudolymphomatous lesions
- ▪
Most common in men over 50 years of age
- ▪
Phototests positive to varying combinations of UVB, UVA, and visible light
- ▪
Positive patch or photopatch tests common
- ▪
Management by photoprotection, avoidance of (photo)contact sensitizers, intermittent oral and topical corticosteroids, topical tacrolimus, low-dose PUVA, cyclosporine, azathioprine, or mycophenolate mofetil
- ▪
Probability of spontaneous resolution is 10% over 5 years, 20% over 10 years
Introduction and history
Chronic actinic dermatitis (CAD) is an uncommon, persistent, often disabling dermatosis of uncovered (and to a lesser extent, covered) skin, generally affecting older men, particularly in the summer. It is evoked by UVR and occasionally also visible light exposure, and CAD probably represents a contact allergy-like DTH response against endogenous photoinduced antigen(s).
Reference to a condition suggestive of CAD was first made in 1933 by Haxthausen, then in the 1960s by Wilkinson in the UK and Jillson and Baughman in the US, with the latter authors terming it “persistent light reaction”. In 1969, its severe form, actinic reticuloid, was described, followed shortly thereafter by reports of two milder variants, photosensitive eczema and photosensitivity dermatitis. In 1979, the current encompassing term, CAD, was introduced by Hawk and Magnus .
Epidemiology
CAD has been regularly observed throughout the world.
Pathogenesis
The action spectra for CAD include: UVB; UVB and UVA; UVB, UVA and visible light; and, rarely, UVA alone, with UVB plus UVA being the most common. The eczematous clinical and histologic appearances of CAD, its dermal infiltrate of primarily CD8 + T lymphocytes, and its pattern of adhesion molecule activation all closely mimic the DTH response of allergic contact dermatitis, strongly suggesting that CAD is presumably a reaction against endogenous, photoinduced, cutaneous antigen(s).
CAD most commonly affects older outdoor workers and enthusiasts, particularly those with a pre-existing allergic or photoallergic contact dermatitis to exogenous sensitizers such as Compositae or sunscreens, respectively . It is therefore possible that in CAD chronic photodamage leads to a partial abrogation of normal cutaneous immunosuppression, thereby permitting enhanced cutaneous immune responsiveness against an otherwise weak endogenous photoantigen. It should be noted that positive patch testing to Compositae, commonly observed in the UK, has not been confirmed in the US and Japan .
It is likely that nucleic acids may be involved as chromophores in the pathogenesis of CAD, in that the shape of the CAD induction spectrum resembles that of sunburn inflammation, for which UVR absorption by nucleic acids is known to be responsible .
Clinical features
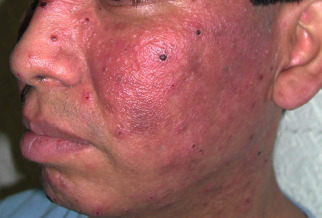
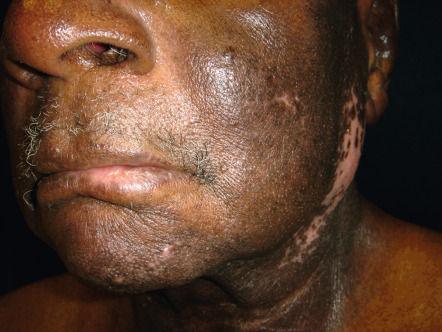
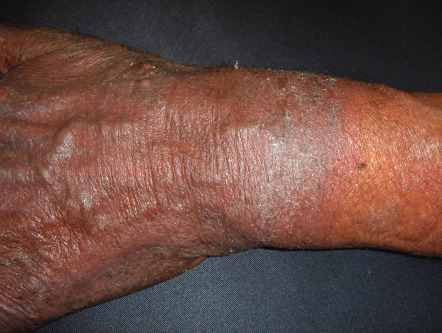
CAD occurs primarily in temperate zones, most commonly affecting older men of any race ; more recently, the disorder has been increasingly recognized in younger women. However, familial incidence has not been noted. The disorder may develop in previously normal skin, in patients with a prior history of dermatitis (in particular, photoallergic or allergic contact dermatitis), or rarely following longstanding oral drug photosensitivity or PMLE. Coexistent allergic contact sensitivity to plant antigens, fragrances or topical medications is common. The CAD eruption is pruritic, patchy or confluent, and the eczematous lesions can be acute, subacute or chronic in nature; the latter is frequently associated with lichenification ( Fig. 87.12 ). Scattered or widespread, erythematous, shiny, infiltrated, pseudolymphomatous papules or plaques may be present in severely affected individuals.
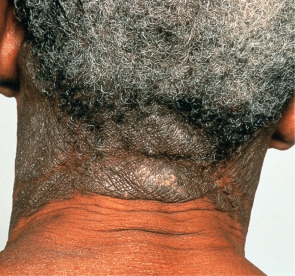
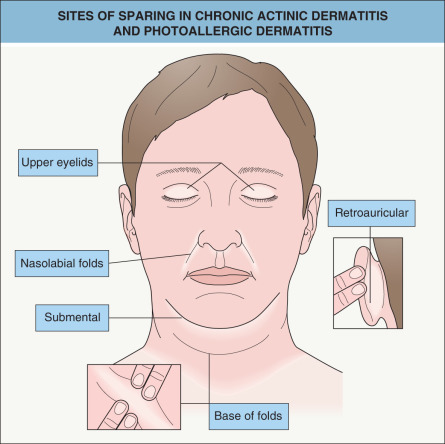
Lesions develop primarily within sun-exposed sites ( Figs 87.13 & 87.14 ), frequently with a sharp cut-off at the lines of clothing, and often with sparing in the depths of skin furrows, upper eyelids, finger webs, nasolabial folds, or postauricular areas ( Fig. 87.15 ). Occasionally, palmoplantar eczematous changes may also be present, while eyebrow or scalp hairs may be stubbly or sparse from rubbing or scratching. Rarely, erythroderma develops in severely affected patients. The probability of resolution has been estimated to be 10% over 5 years, 20% over 10 years, and 50% over 15 years .
Pathology
There is epidermal spongiosis and acanthosis, lymphocytic exocytosis, and a superficial and deep, often dense, perivascular lymphohistiocytic dermal cellular infiltrate, often containing eosinophils and plasma cells . In more severely affected skin, erosions, focal epidermal necrosis, Pautrier microabscess-like epidermal cellular collections, dermal–epidermal junction fibrin deposition, dermal neutrophils with nuclear dust, vertically streaked papillary dermal collagen, and/or small, multinucleated dermal giant cells are often seen; in addition, the dense infiltrate, epidermal lymphocyte exocytosis, and frequent nuclear polymorphism may on occasion suggest a histologic diagnosis of cutaneous T-cell lymphoma (CTCL).
Differential diagnosis
Photoaggravated dermatoses are differentiated by the characteristic clinical features of the underlying primary disorder and normal cutaneous irradiation responses. In drug- and chemical-induced photosensitivity, there is usually a history of exposure to a relevant drug or allergen or clinical evidence (e.g. positive photopatch testing results; see below).
In contrast to CTCL, CAD infiltrates are predominantly CD8 + cells and T-cell receptor gene rearrangement studies are negative. Very rarely, primary CTCL may present with severe CAD features and should therefore be considered if apparent CAD is totally refractory to treatment . Erythrodermic CAD must be differentiated from other causes of erythroderma (see Ch. 10 ), but histologic features may not allow discrimination so phototesting after the erythroderma has been controlled can assist in establishing the diagnosis.
Treatment
Strict photoprotection and the avoidance of relevant contact allergens are of primary importance. Of note, computer and video screens are safe. Commercially available films that block UVR can be placed on home and car windows. Topical or intermittent oral corticosteroid therapy along with emollient use is generally needed, and topical tacrolimus may also be helpful . Therapy for refractory disease includes very low-dose PUVA with initial high-dose oral and topical corticosteroid coverage over months, cyclosporine (3.5–5 mg/kg/day), azathioprine (1.0–2.5 mg/kg/day if normal TPMT activity), or mycophenolate mofetil (1–2 g/day).
Solar urticaria
- ▪
A physical urticaria induced by sun exposure; lesions appear within 5 to 10 minutes and resolve over 1 to 2 hours
- ▪
The action spectrum generally includes both UVA and visible light, but rarely UVB
- ▪
There is a slight female predominance, often with an onset during the third decade of life
- ▪
Estimated probability of resolution is 15% at 5 years, 25% at 10 years
- ▪
Management: photoprotection; high-dose, non-sedating antihistamines; low-dose UVA; PUVA; plasmapheresis; IVIg; and omalizumab
Introduction and history
In solar urticaria, transient wheals appear within 5 to 10 minutes of exposure to UV or visible radiation. Lesions then resolve over 1 to 2 hours. Solar urticaria was first reported in 1905.
Epidemiology
Solar urticaria is a relatively uncommon disorder; one study from Scotland reported a prevalence of ~3.1 per 100 000 . There is a slight female predominance, and the onset is most frequently during the third decade of life. Solar urticaria may occasionally coexist with PMLE, CAD, and very rarely, EPP .
Pathogenesis
As in other forms of urticaria (see Ch. 18 ), mast cells play a major role. Although factors triggering their degranulation have not been fully elucidated, an IgE-mediated response against photoinduced, endogenous cutaneous antigen(s) is a likely mechanism. This is supported by observations that whealing may be induced by previously in vitro -irradiated injections of autologous serum, and then blocked by anti-IgE antibody-containing serum. Also, in some patients, plasmapheresis is an effective treatment.
Clinical features
Patients present with whealing limited to sun-exposed areas, particularly the upper chest and extensor arms, within 5 to 10 minutes of sun exposure. Occasionally, erythema without urtication may occur. The lesions then resolve over 1 to 2 hours. Patients almost invariably note an accompanying pruritus, burning sensation, or rarely pain, while severe attacks are rarely associated with malaise, light-headedness, nausea, bronchospasm, and/or syncope.
Although solar urticaria is oftentimes classified by its action spectrum, in an individual patient, the spectrum may gradually change over time . Most commonly, visible light is responsible, but usually with extension into the UVA waveband. In some patients, the action spectrum may include UVB .
Occasionally, continued regular skin exposure to UVR may decrease the likelihood of attacks of solar urticaria, a phenomenon referred to as hardening. The probability of spontaneous resolution has been estimated as being 15% at 5 years and 25% at 10 years after onset .
There are several uncommon manifestations of this disorder. Fixed solar urticaria is limited to specific areas, presumably due to mast cell alterations just at those sites . “Drug-induced” solar urticaria has been reported following exposure to chlorpromazine, tetracycline, or tar. Solar urticaria has also been observed after light-emitting diode therapy.
Pathology
The histologic findings are similar to those of other urticarias. There is mild dermal edema with a sparse perivascular mixed neutrophilic and eosinophilic infiltrate; mononuclear cells may also be seen. In addition, eosinophil major basic protein has been demonstrated within the dermis by immunofluorescence.
Differential diagnosis
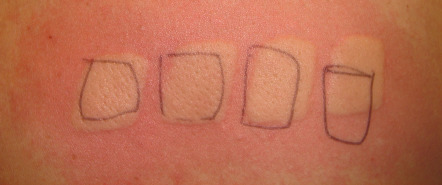
The differential diagnosis includes other forms of urticaria, particularly heat urticaria, PMLE, and very rarely, EPP. A detailed history should differentiate solar urticaria from idiopathic urticaria and other physical urticarias. Phototesting of affected patients generally results in typical whealing within minutes ( Fig. 87.16 ), while the use of a water filter in front of the light source eliminates heat to exclude heat urticaria. The rare association of solar urticaria with EPP can be excluded by determination of RBC protoporphyrins (see Ch. 49 ). The time course of PMLE is distinct from that of solar urticaria, and LE can be excluded via laboratory evaluation, if the history and physical examination are inconclusive (see Table 87.3 ).
Treatment
Oral non-sedating antihistamines, often in high doses and taken regularly or about an hour or so before expected sun exposure, are effective in ~50% of patients. Those with very mild disease may occasionally require only photoprotective measures; however, sunscreens do not generally provide sufficient protection against UVA and visible light to be effective . Graduated exposures to UVA or PUVA are occasionally helpful , and some patients have experienced a beneficial effect for years. If these measures fail, more aggressive therapies such as omalizumab (anti-IgE antibody), plasmapheresis , or IVIg can be tried.
Photosensitity Due to Hereditary Defects in DNA Repair
The major disorders characterized by both defective DNA repair and photosensitivity are discussed below and outlined in Table 87.4 . The underlying pathogenesis is reviewed in greater detail in Chapter 86 . Five of the disorders are associated with defects in one of the nucleotide excision repair (NER) proteins: xeroderma pigmentosum, Cockayne syndrome, cerebro-oculo-facio-skeletal (COFS) syndrome, UV-sensitive syndrome, and the photosensitive form of trichothiodystrophy .
| INHERITED PHOTOSENSITIVITY DISORDERS ASSOCIATED WITH DEFECTIVE DNA NUCLEOTIDE EXCISION REPAIR OR CHROMOSOME INSTABILITY | |||||
|---|---|---|---|---|---|
| Disorder | Clinical features | Inheritance | Photosensitivity | Action spectrum | Laboratory findings |
| Xeroderma pigmentosum (XP) |
| AR | Yes | 290–340 nm |
|
| Cockayne syndrome (CS) |
| AR | Yes | UVB, UVA |
|
| Cerebro-oculo-facio-skeletal (COFS) syndrome |
| AR | Yes | UVB, UVA |
|
| UV-sensitive syndrome (UV s S) |
| AR | Yes | Unclear |
|
| Trichothiodystrophy (TTD) |
| AR | Yes (in ~50% of patients) TTD1, 2, 3 | Unclear |
|
| Bloom syndrome |
| AR | Yes | Unclear |
|
| Rothmund–Thomson syndrome |
| AR | Transient (early childhood) | UVA |
|
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


