Abstract
The perforating diseases are a group of disorders in which papulonodules develop due to transepidermal elimination of connective tissue or other substances. Two prototypes are reactive perforating collagenosis (RPC), in which collagen is the primary extruded material, and elastosis perforans serpiginosa (EPS), in which elastic tissue is involved. RPC and EPS may be inherited, but are more often acquired and related to an inciting condition. Kyrle disease is a third form of perforating disease and is related to perforation of nonspecific connective tissue; it is closely related to prurigo nodularis. Collectively, acquired perforating dermatosis, regardless of any subclassification as acquired RPC or Kyrle disease, is most often related to diabetes mellitus and/or chronic kidney disease (stages 4–5), with pruritus leading to prurigo lesions and connective tissue perforation through the epidermis. EPS can be related to long-term penicillamine administration as well as a number of genetic disorders, e.g. Marfan syndrome, Ehlers–Danlos syndrome. Keratotic plugs are common to all perforating disorders, both clinically and histologically. Lesions are often arranged in an annular pattern in EPS while a linear pattern due to the Koebner phenomenon is most commonly observed in RPC. Perforating folliculitis is often cited as the fourth major perforating disease, but in reality it is no different than any generic folliculitis in which the hair follicle has ruptured. Perforating disorders are difficult to treat, and various destructive methods are most commonly employed, such as curettage or cryotherapy. When there is extensive disease, phototherapy may be helpful.
Keywords
perforating disease, perforating disorder, reactive perforating collagenosis, RPC, elastosis perforans serpiginosa, EPS, acquired reactive perforating collagenosis, acquired perforating dermatosis, perforating folliculitis, Kyrle disease, transepidermal elimination, diabetes mellitus, chronic renal disease, penicillamine, perforating calcific elastosis
- ▪
Group of disorders with transepidermal elimination of collagen, elastic tissue, or necrotic dermal connective tissue
- ▪
Papules or nodules with keratotic plugs
- ▪
Elastosis perforans serpiginosa is associated with genetic diseases or penicillamine administration and involves elastic tissue; lesions are typically annular and most commonly occur in flexural sites, in particular the neck
- ▪
Reactive perforating collagenosis occurs after minor trauma, involves collagen, and often involves the upper extremities
- ▪
Acquired perforating dermatosis is almost always associated with diabetes mellitus and/or the pruritus of chronic kidney disease (stages 4–5); it favors the lower extremities of adults
- ▪
Broadband or narrowband UVB phototherapy appears to be the most effective treatment for acquired perforating dermatosis
Introduction
The perforating diseases are a group of papulonodular skin disorders characterized by keratotic plugs or crusts in which dermal connective tissue “perforates” or is eliminated through the epidermis ( Table 96.1 ). There are two prototypic perforating diseases, both of which can be inherited. Reactive perforating collagenosis (RPC) is associated with transepidermal elimination of primarily collagen fibers , whereas in elastosis perforans serpiginosa (EPS), primarily elastic fibers perforate the epidermis. The third major perforating disease is acquired perforating dermatosis , which usually develops during adulthood in association with diabetes mellitus and/or the pruritus of chronic kidney disease (stages 4–5) . Many textbooks traditionally list “perforating folliculitis” as the fourth perforating disease , but, in the opinion of the author, this seems unjustified. It does not appear to be a specific entity, since perforation or rupture of follicles occurs in a wide variety of diseases classified as folliculitis, regardless of whether the pathogenesis involves bacteria, fungi, Demodex mites, drugs, physical trauma, or other mechanisms.
| MAJOR PERFORATING DISEASES | |||||
|---|---|---|---|---|---|
| Disease | Incidence | Time of onset | Most common location | Perforating substance | Associations |
| Familial reactive perforating collagenosis (RPC) | Very rare | Childhood | Arms, hands, sites of trauma | Collagen | None |
| Elastosis perforans serpiginosa (EPS) | Rare, M > F | Childhood, early adulthood; variable with penicillamine-induced EPS | Neck, face, arms, other flexural areas | Elastic tissue | Genetic diseases (see Fig. 96.13 ), penicillamine |
| Perforating folliculitis | Common | Early adulthood | Trunk, extremities | Necrotic material | May simply be ordinary folliculitis with follicular rupture, i.e. not a specific entity |
| Acquired perforating dermatosis, includes acquired RPC, Kyrle disease * , and rarely, acquired EPS | Common (10% of dialysis patients) | Adulthood | Legs or generalized | Necrotic material, collagen, or uncommonly, elastic tissue | Diabetes, chronic kidney disease (stages 4–5), pruritus, rarely liver disease; may be end stage of perforating folliculitis |
| Perforating calcific elastosis | Very rare, more common in black women | Adulthood | Abdomen, periumbilical | Calcified elastic tissue | Multiparity, weight gain, obesity, hypertension |
Some authorities have expanded the concept of the preceding so-called “primary” perforating diseases to include a variety of unrelated “secondary” perforating disorders in which transepidermal elimination of a substance occurs as a secondary component of a primary dermatosis ( Table 96.2 ). As in most of the preceding perforating disorders, the epidermis often becomes hyperplastic, eventually surrounds the material to be extruded, and subsequently causes the material’s elimination via normal keratinocyte maturation. Some of these disorders are discussed elsewhere in this text and involve perforation of endogenous substances, exogenous foreign material, infectious organisms, granulomas, and even neoplastic cells .
| SECONDARY PERFORATING DISEASES |
| Endogenous substances |
|
| Granulomas |
|
| Exogenous foreign material |
|
| Infectious diseases |
|
| Tumor cells |
|
History
In 1916, Kyrle reported a diabetic woman with generalized hyperkeratotic nodules, which he called “hyperkeratosis follicularis et parafollicularis in cutem penetrans”. Lutz first described EPS as “keratosis follicularis serpiginosa” in 1953, and, in 1955, Miescher described the histologic findings, naming it “elastoma intrapapillare perforans verruciforme”. Mehregan, Schwartz and Livingood reported the first child with RPC in 1967. A year later, Mehregan and Coskey expanded the concept of perforating disease to include perforating folliculitis in their article.
Epidemiology
The perforating diseases are found worldwide, without any clear racial predilection. The very rare childhood-onset form of RPC is commonly familial, and childhood-onset EPS is occasionally familial. Although the exact inheritance pattern for both is still uncertain, isolated inherited EPS may have an autosomal dominant pattern. EPS affects men more often than women, with a ratio of ~4 : 1, whereas the gender distribution is nearly equal in RPC and acquired perforating dermatosis. Perforating folliculitis is said to be more common in women. In patients receiving maintenance hemodialysis, acquired perforating dermatosis is observed in an estimated 4.5% to 11% .
Pathogenesis
The precise pathogenesis underlying the perforating disorders is unknown. As it is unlikely that dermal connective tissue or hair shafts actively perforate into the epithelium, some have argued that “transepidermal elimination” is a more accurate term. The term “perforating” is embedded in the dermatologic lexicon, however, and is easier to say. Epithelium becomes hyperplastic and eventually surrounds the abnormal connective tissue, just as it appears to do with wood splinters or other foreign bodies. In acquired perforating dermatosis, pruritus probably leads to chronic scratching, which results in epithelial hyperplasia, as in prurigo nodularis. Indeed, prurigo nodules are often admixed with classic perforating lesions. The possibility has been raised that enlargement of a hyperkeratotic plug could lead to penetration through the base of the hyperplastic epidermis, i.e. the perforation is caused by the plug above rather than being due to the abnormal connective tissue below.
Primary perforating diseases may be due to either genetic or acquired abnormalities of collagen or elastic fibers. In EPS, as well as some cases of acquired perforating dermatosis in which perforation of elastic fibers is observed, enhanced expression of elastin receptors has been detected in the epidermis surrounding the elastic material . Recently, it was proposed that scratching exposes keratinocytes to a dvanced g lycation e nd product (AGE)-modified extracellular matrix proteins, in particular collagen types I and III. This would then lead to terminal differentiation of keratinocytes via the AGE receptor (CD36), followed by upward movement of keratinocytes along with glycated collagen .
Plasma fibronectin levels are elevated in patients with diabetes mellitus and uremia, and fibronectin has also been found to be increased within the skin at sites of transepidermal elimination . This may be significant, since fibronectin plays a role in epithelial cell signaling, locomotion, and differentiation. It binds to type IV collagen (the type found in basement membranes) and to keratinocytes, and may incite epithelial proliferation and perforation. In one study of RPC, the investigators demonstrated that the collagen being transepidermally eliminated was type IV collagen . Increased expression of transforming growth factor-β 3 (TGF-β 3 ), matrix metalloproteinase-1 (MMP-1), and tissue inhibitor of metalloproteinase-1 (TIMP-1) has been observed in lesional skin of acquired RPC , but this enhanced expression may be a reflection of concurrent wound healing (see Ch. 141 ).
Other proposed pathomechanisms include abnormal vitamin A or D metabolism, enzyme release from neutrophils , and microangiopathy related to diabetes. Deposition of uric acid, hydroxyapatite, or silicon has also been implicated in the pathogenesis of perforating disorders.
Clinical Features
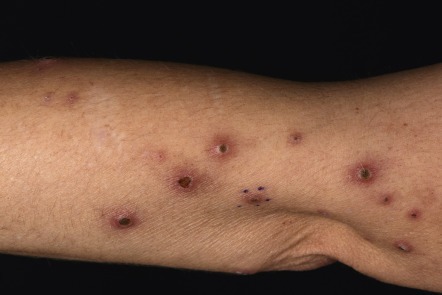
Reactive Perforating Collagenosis
Familial RPC is a rare disorder that begins during childhood . After superficial trauma, patients develop keratotic papules that reach a size of 5–8 mm over the following 3–4 weeks. The Koebner phenomenon may occur, in which injury to the skin results in the formation of new lesions, often in a linear distribution. Koebnerization is more commonly observed with RPC than with the other perforating disorders, but it has been reported with all of them. Arms and hands are the most common sites of involvement in RPC ( Fig. 96.1 ). The papules tend to spontaneously resolve over 6–10 weeks. Verrucous perforating collagenoma ( collagenome perforant verruciformé ) is a very rare non-familial variant of RPC in which severe trauma to the skin results in verrucous papules with transepidermal elimination of collagen . Acquired RPC that begins in adulthood usually occurs in association with diabetes mellitus and/or chronic kidney disease (stages 4–5), and these cases are best classified as acquired perforating dermatosis, even though the histopathology can be identical to the inherited form of RPC.