Abstract
Pemphigus is a group of IgG autoantibody-mediated blistering diseases of the skin and mucous membranes that includes three major forms: pemphigus vulgaris, pemphigus foliaceus, and paraneoplastic pemphigus. Histologically, there is intraepidermal blister formation due to the loss of cell–cell adhesion of keratinocytes. Immunopathologic studies serve to identify in vivo bound and circulating IgG autoantibodies against desmogleins found within desmosomes. Patients with pemphigus vulgaris and pemphigus foliaceus have IgG autoantibodies against desmoglein 3 and desmoglein 1, respectively, while patients with paraneoplastic pemphigus also have IgG autoantibodies against plakin molecules as well as a T-cell-mediated autoimmune reaction that leads to an interface dermatitis. Systemic corticosteroids are a mainstay of therapy, but due to their toxicity at effective doses, immunosuppressive medications are regularly used as steroid-sparing agents. More recently, high-dose IVIg, which is non-immunosuppressive, and rituximab, a B-cell-depleting monoclonal antibody, have been added to the therapeutic armamentarium for pemphigus.
Keywords
autoantibody, autoimmune bullous disease, cell adhesion, desmosomes, desmoglein, cadherin, blister, acantholysis, pemphigus vulgaris, pemphigus, pemphigus foliaceus, pemphigus erythematosus, fogo selvagem, pemphigus vegetans, IgA pemphigus, drug-induced pemphigus, paraneoplastic pemphigus
▪ Fogo selvagem: endemic pemphigus foliaceus; Brazilian pemphigus ▪ Pemphigus erythematosus: Senear–Usher syndrome ▪ IgA pemphigus: intercellular IgA dermatosis; intraepidermal neutrophilic IgA dermatosis; intercellular IgA vesiculopustular dermatosis ▪ Paraneoplastic pemphigus: paraneoplastic autoimmune multi-organ syndrome
- ▪
Pemphigus is a group of autoimmune blistering diseases of the skin and mucous membranes that is characterized by:
- •
histologically, intraepidermal blisters due to the loss of cell–cell adhesion of keratinocytes
- •
immunopathologically, the finding of in vivo bound and circulating IgG autoantibodies directed against the cell surface of keratinocytes
- •
- ▪
Pemphigus is divided into three major forms: pemphigus vulgaris, pemphigus foliaceus, and paraneoplastic pemphigus
- ▪
The functional inhibition of desmogleins, which play an important role in cell–cell adhesion of keratinocytes, by IgG autoantibodies results in blister formation
- ▪
Patients with pemphigus vulgaris and pemphigus foliaceus have IgG autoantibodies against desmoglein 3 and desmoglein 1, respectively, while patients with paraneoplastic pemphigus also have IgG autoantibodies against plakin molecules as well as a T-cell-mediated autoimmune reaction that leads to an interface dermatitis
- ▪
IgA pemphigus is characterized by IgA, but not IgG, autoantibodies directed against keratinocyte cell surfaces and is divided into two major subtypes: intraepidermal neutrophilic (IEN) type and subcorneal pustular dermatosis (SPD) type
- ▪
Systemic corticosteroids are a mainstay of therapy in pemphigus vulgaris, given the rapidity of clinical response, but because of their potential side effects at effective doses, they are combined with steroid-sparing agents
- ▪
These additional therapies include immunosuppressive medications such as mycophenolate mofetil, high-dose IVIg (non-immunosuppressive), and rituximab; in the future, the latter may become a first-line therapy
Introduction
The term pemphigus stems from the Greek pemphix , meaning blister or bubble, and it describes a group of chronic blistering skin diseases in which autoantibodies are directed against the cell surface of keratinocytes, resulting in the loss of cell–cell adhesion of keratinocytes through a process called acantholysis . Pemphigus can be divided into three major forms: pemphigus vulgaris, pemphigus foliaceus, and paraneoplastic pemphigus ( Table 29.1 ).
| CLASSIFICATION OF PEMPHIGUS |
|
Pemphigus vulgaris and pemphigus foliaceus are the originally characterized, classic forms of pemphigus. All patients with pemphigus vulgaris have mucosal membrane erosions, and more than half will also have cutaneous blisters and erosions. The blisters of pemphigus vulgaris develop within the deeper portion of the epidermis, just above the basal cell layer. Patients with pemphigus foliaceus only have cutaneous involvement without mucosal lesions, and the splits occur in the superficial portion of the epidermis, mostly at the granular layer. Pemphigus vegetans is a variant of pemphigus vulgaris, and pemphigus erythematosus and fogo selvagem represent localized and endemic variants of pemphigus foliaceus, respectively.
Paraneoplastic pemphigus is distinct from the classic forms of pemphigus , as it consists of both humoral and cellular autoimmune reactions. Patients with paraneoplastic pemphigus have a known or occult associated neoplasm, usually of lymphoid tissue. Painful, severe oral and conjunctival erosions are a prominent feature of paraneoplastic pemphigus.
History
The modern history of pemphigus began with the discovery by Beutner and Jordon in 1964 of circulating antibodies directed against the cell surface of keratinocytes in the sera of pemphigus vulgaris patients ( Fig. 29.1 ). This was followed by the finding of in vivo IgG deposition on the cell surface of keratinocytes in patients’ skin. These discoveries formed the basis of our understanding of pemphigus as a tissue-specific autoimmune disease of the skin and mucous membranes. In the late 1970s and early 1980s, pemphigus autoantibodies were shown to be pathogenic, i.e. they could induce blister formation in skin organ-culture systems as well as by passive transfer of patients’ IgG to neonatal mice . In the mid and late 1980s, the target antigens of pemphigus were characterized by immunochemical methods, such as immunoprecipitation and immunoblotting . In the early 1990s, the isolation of cDNA for pemphigus antigens demonstrated that pemphigus is an anti-cadherin autoimmune disease .
Epidemiology
The prevalence of pemphigus vulgaris and pemphigus foliaceus in men and women is approximately equal. The mean age of onset of disease is 50 to 60 years, although the range is broad and disease arising in the elderly and in children has been described. Pemphigus can be found all over the world. Although limited data are available regarding the incidence of pemphigus, in general, it ranges from 0.76 to 5 new cases per million per year. However, the incidence is much higher (16 to 32 cases per million per year) in those of Jewish ancestry. Occasionally, this ancestry may not be readily apparent, as in the Hispanic descendants of the conversos , who currently reside in the southwestern USA.
In most countries, pemphigus vulgaris is more common than pemphigus foliaceus; exceptions include Finland, Tunisia and Brazil. For example, in Japan (incidence of pemphigus: 3.5 cases per million per year), the ratio of pemphigus vulgaris to pemphigus foliaceus is 2 : 1. In France (incidence: 1.7 cases per million per year), pemphigus vulgaris accounts for the majority of all cases (~75%), as it does in Bulgaria. In contrast, in Finland (incidence: much lower at 0.76 cases per million per year) and in Tunisia (incidence: 6.7 cases per million per year), pemphigus foliaceus is twice as common as pemphigus vulgaris. More interestingly, the female-to-male ratio in Tunisia is 4 : 1, and the incidence is higher in young women ages 25 to 34 years (15.5 cases per million per year). In addition to Brazil, Tunisia may have endemic foci of pemphigus foliaceus as may the Mt. Kilimanjaro area.
Fogo Selvagem
Patients with fogo selvagem are clinically, histologically and immunopathologically similar to patients with sporadic pemphigus foliaceus. However, fogo selvagem occurs in an endemic fashion in certain regions of Brazil and is thought to be caused by an environmental factor(s). Unlike sporadic pemphigus foliaceus, which is a disease of mostly middle-aged and older patients, fogo selvagem affects young adults and children of either sex or any race who are exposed to the local ecology in rural areas, and the incidence gradually decreases as the area is developed. The majority of patients live close to rivers and within the 10–15 km flying range of black flies ( Simulium spp.), which may be the vector that precipitates the disease. There is also a high frequency of hematophagous insects (bedbugs and kissing bugs) in the homes of patients with fogo selvagem.
Fogo selvagem frequently occurs in genetically related family members, although it is not contagious, and, to date, spread by blood products or body fluids has not been documented. In rural Brazil, the ratio of pemphigus foliaceus to pemphigus vulgaris is a remarkable 17 : 1. The prevalence in some rural areas of Brazil is as high as 3.4% of the population . Furthermore, in these areas, more than 50% of normal individuals have anti-desmoglein 1 (anti-Dsg1) IgG autoantibodies, and it has been shown that the onset of the disease is preceded by a sustained antibody response consisting of non-pathogenic IgG1 and IgG4 antibodies. In certain genetically predisposed individuals, predominant IgG4 antibodies directed against the N-terminal extracellular EC1 and EC2 domains of Dsg1 are responsible for inducing disease ( Fig. 29.2 ) . Certainly, fogo selvagem provides a fascinating model for understanding the pathophysiologic and immunologic mechanisms involved in triggering an autoimmune response against skin components.
Pathogenesis
Pathogenic Autoantibodies in Pemphigus
The hallmark of pemphigus is the finding of IgG autoantibodies against the cell surface of keratinocytes (see Fig. 29.1 ) . The pemphigus autoantibodies found in patients’ sera play a primary pathogenic role in inducing the loss of cell adhesions between keratinocytes, and subsequent blister formation. Neonates of mothers with pemphigus vulgaris may have a transient disease caused by maternal IgG that crosses the placenta. As maternal antibody is catabolized, the disease subsides. IgG fractions from patients can induce blister formation in the absence of complement or inflammatory cells in a skin organ-culture system . Furthermore, passive transfer of patients’ IgG to neonatal mice results in blisters in the mice with typical histologic findings . Even monovalent Fab′ fragments of IgG from patients with pemphigus foliaceus are sufficient to cause blisters in neonatal mice, indicating complement activation and surface cross-linking may not be essential in keratinocyte detachment .
Desmogleins as Pemphigus Antigens
Immunoelectron microscopy localized both pemphigus vulgaris and pemphigus foliaceus antigens to the desmosomes, the most prominent cell–cell adhesion junctions in stratified squamous epithelia . Immunochemical characterization of pemphigus antigens by immunoprecipitation or immunoblotting with extracts from cultured keratinocytes or epidermis demonstrated that the pemphigus vulgaris and foliaceus antigens were 130 kDa and 160 kDa transmembrane glycoproteins, respectively ( Table 29.2 ) . By comparative immunochemical studies using anti-desmoglein 1 (Dsg1) monoclonal and polyclonal antibodies, the 160 kDa protein recognized by pemphigus foliaceus sera was subsequently shown to be identical to Dsg1. An 85 kDa plaque protein, plakoglobin, was co-immunoprecipitated with the 130 kDa and 160 kDa pemphigus antigens, demonstrating that plakoglobin forms a molecular complex with pemphigus vulgaris and foliaceus antigens .
| TARGET ANTIGENS IN PEMPHIGUS | |||
|---|---|---|---|
| Disease | Autoantibodies | Antigens | MW (kDa) |
| Pemphigus vulgaris | |||
| Mucosal-dominant type | IgG | Desmoglein 3 | 130 |
| Mucocutaneous type | IgG | Desmoglein 3 | 130 |
| Desmoglein 1 | 160 | ||
| Pemphigus foliaceus | IgG | Desmoglein 1 | 160 |
| Paraneoplastic pemphigus | IgG | Desmoglein 3 | 130 |
| Desmoglein 1 | 160 | ||
| Plectin * | 500 | ||
| Epiplakin * | 500 | ||
| Desmoplakin I * | 250 | ||
| Desmoplakin II * | 210 | ||
| BPAG1 * | 230 | ||
| Envoplakin * | 210 | ||
| Periplakin * | 190 | ||
| A2ML1 | 170 | ||
| Drug-induced pemphigus | IgG | Desmoglein 3 | 130 |
| Desmoglein 1 | 160 | ||
| IgA pemphigus † | |||
| Subcorneal pustular dermatosis type | IgA | Desmocollin 1 | 110/100 |
| Intraepidermal neutrophilic type | IgA | ? | ? |
† A subset of patients have IgA autoantibodies against Dsg1 or Dsg3.
Molecular cloning of cDNA encoding Dsg1 and pemphigus vulgaris antigens indicated that both molecules were members of the cadherin supergene family . Thus, pemphigus was discovered to be an anti-cadherin autoimmune disease. The pemphigus vulgaris antigen was termed desmoglein 3 (Dsg3). The basic pathophysiology of pemphigus is as follows: autoantibodies inhibit the adhesive function of desmogleins and lead to the loss of the cell–cell adhesion of keratinocytes, resulting in blister formation.
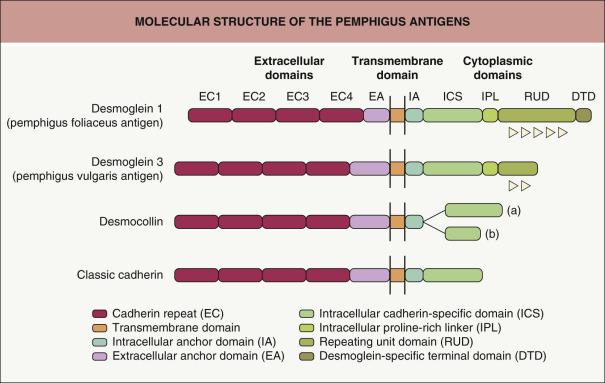
Cadherins are a family of calcium-dependent cell–cell adhesion molecules that play an important role in the formation and maintenance of complex tissue integrity . Based on sequence similarity, cadherins have two major subgroups, classic cadherins (e.g. E-, P-, N-cadherins) and desmosomal cadherins (desmogleins and desmocollins). All members of the cadherin family contain conserved repeated amino acid sequences (cadherin repeats) with calcium-binding motifs in their extracellular domains (see Fig. 29.2 ). When classic cadherins were introduced by gene transfection into non-adhesive mouse fibroblast L cells, the cells acquired strong cell adhesion activity mediated by a homophilic type of interaction. Cadherins require their well-conserved cytoplasmic domains in order to associate with the plaque proteins, α-catenin, β-catenin and plakoglobin, which mediate and regulate binding to the cytoskeleton network ( Fig. 29.3 ). As a consequence of these interactions, cadherins produce strong cellular adhesion and morphologic changes in the cells. Cadherin molecules form dimers as their functional unit, with the distal extracellular domain (EC1) of the cadherin from one cell binding to the same region of a second cadherin from an opposing cell .
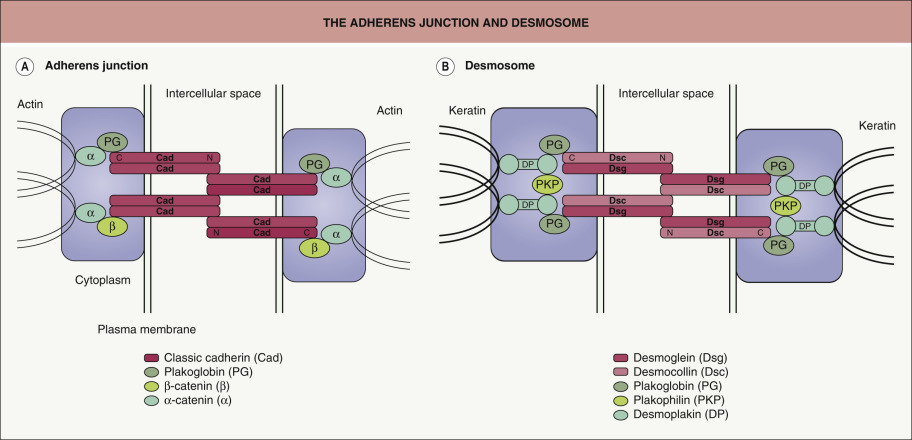
Two major types of adhering junctions of epithelial cells are commonly distinguished: adherens junctions and desmosomes (see Fig. 29.3 ) . The adherens junction anchors bundles of actin microfilaments and contains classic cadherins as its transmembrane components and α-catenin, β-catenin and plakoglobin as its cytoplasmic components. In contrast, the desmosome anchors intermediate filaments like keratins and contains desmosomal cadherins as its transmembrane components and plakoglobin, plakophilin and desmoplakin as its cytoplasmic components. In general, adherens junctions mediate quick but weak cellular adhesion, whereas desmosomes mediate slow but strong cellular adhesion.
Desmogleins have four cadherin repeats in their extracellular domain, as do classic cadherins (see Fig. 29.2 ). Desmogleins have four isoforms (Dsg1–4). Expression of Dsg1 and Dsg3 is basically restricted to stratified squamous epithelia, where blisters are formed in pemphigus, while Dsg2 is expressed in all desmosome-possessing tissues, including simple epithelia and myocardium. Dsg4 plays an important adhesive role primarily in hair follicles, and mutations in the DSG4 gene can lead to abnormal hair development (e.g. localized autosomal recessive hypotrichosis) .
Desmocollins are another group of transmembrane glycoproteins within desmosomes and they have three isoforms (Dsc1–3). Each isoform has two products derived from alternatively spliced mRNA of a single gene. Desmosomes always have desmoglein and desmocollin as a pair, but the exact molecular fashion of their interaction as well as the reason why desmocollin does not compensate for the loss of desmoglein in pemphigus remain to be elucidated.
Plakoglobin and plakophilin, together with β-catenin, are members of the armadillo family of nuclear and junctional proteins, which are not only simple anchoring molecules but also dynamic regulators of cellular adhesion and proliferation. Desmoplakin is a dumbbell-shaped molecule composed of three domains: a central α-helical coiled-coil rod, flanked by globular carboxy- and amino-terminal domains that interact with intermediate filaments and armadillo family members, respectively (see Fig. 29.3 ). Desmoplakin has two products derived from alternatively spliced mRNA of a single gene: desmoplakin I (250 kDa) and II (210 kDa) (see Table 29.2 ). Desmoplakin, a member of the plakin family, plays an important role in the anchorage of the cytoskeleton to filament attachment sites on desmosomes.
Compelling evidence has accumulated that IgG autoantibodies against Dsg1 and Dsg3 are pathogenic and play a primary role in inducing the blister formation in pemphigus. Essentially, all patients with pemphigus have IgG autoantibodies against Dsg1 and/or Dsg3, depending on the subtype of pemphigus . When anti-desmoglein IgG autoantibodies are removed from the sera of patients with pemphigus vulgaris, pemphigus foliaceus or paraneoplastic pemphigus (by immunoadsorption with recombinant desmoglein proteins), the sera are no longer pathogenic in inducing blister formation . Furthermore, anti-desmoglein IgG autoantibodies that have been affinity-purified from pemphigus sera via desmoglein recombinant proteins can cause blisters when injected into neonatal mice . Some pemphigus sera react with Dsg4 due to cross-reactivity of a subset of anti-Dsg1 IgG, although Dsg4/Dsg1-cross-reacting IgG has no demonstrable pathogenic effect . IgG autoantibodies against non-desmoglein molecules, such as acetylcholine receptors or annexin-like molecules, have also been detected, but their pathogenic relevance in pemphigus remains to be determined.
Desmoglein Compensation Theory as Explanation for Localization of Blisters
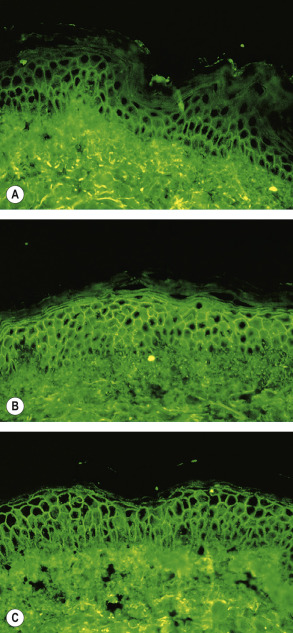
The sites of blisters in pemphigus vulgaris and foliaceus are explained logically by the desmoglein compensation theory: Dsg1 and Dsg3 compensate for each other when they are coexpressed in the same cell ( Fig. 29.4 ) . While patients with pemphigus foliaceus have only anti-Dsg1 IgG autoantibodies, individuals with the mucosal-dominant type of pemphigus vulgaris have only anti-Dsg3 IgG autoantibodies. Those with the mucocutaneous type of pemphigus vulgaris have both anti-Dsg3 and anti-Dsg1 IgG autoantibodies . Of note, the intraepithelial expression pattern of Dsg1 and Dsg3 differs between the skin and mucous membranes. In the skin, Dsg1 is expressed throughout the epidermis, but more intensely in the superficial layers (see Fig. 29.1C ), whereas Dsg3 is expressed in the lower portion of the epidermis, primarily in the basal and parabasal layers (see Fig. 29.1A ). In contrast to the skin, Dsg1 and Dsg3 are expressed throughout the squamous layer of mucosa, but Dsg1 is expressed at a much lower level than Dsg3 (see Fig. 29.4B ) .
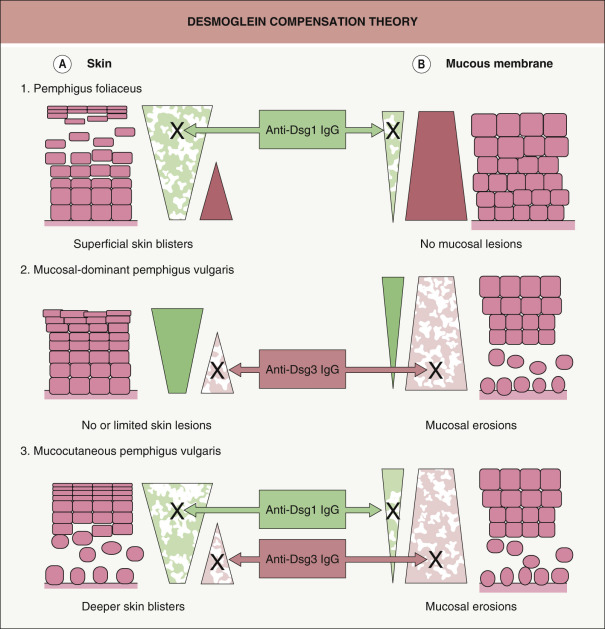
When sera contain only anti-Dsg1 IgG (which interferes with the function of Dsg1), blisters appear only in the superficial epidermis of the skin because that is the only area in which Dsg1 is present without coexpression of Dsg3. In the unaffected deep epidermis, the presence of Dsg3 compensates for the loss of function of Dsg1. Although the anti-Dsg1 IgG binds to mucosa, no blisters are formed, because of the coexpression of Dsg3. Thus, sera containing only anti-Dsg1 IgG cause superficial blisters in the skin without mucosal involvement, as is seen in patients with pemphigus foliaceus.
When sera contain only anti-Dsg3 IgG, they are inefficient in producing cutaneous blisters because coexpressed Dsg1 compensates for the impaired function of Dsg3, resulting in no, or only limited, skin lesions. However, in the mucous membranes, Dsg1 cannot compensate for the impaired Dsg3 function because of its low expression. Therefore, sera containing only anti-Dsg3 IgG cause oral erosions without apparent skin involvement, as is seen in patients with the mucosal-dominant type of pemphigus vulgaris.
When sera contain both anti-Dsg1 and anti-Dsg3 IgG, they interfere with the function of both Dsg1 and Dsg3, resulting in extensive blisters and erosions of the skin as well as the mucous membranes, as is seen in patients with the mucocutaneous type of pemphigus vulgaris. It is not clear why splits appear just above the basal layer instead of the whole epithelium falling apart. However, it is speculated that cell–cell adhesion in the basal and parabasal layers might be weaker than in other parts of the epithelium because there are fewer desmosomes. In addition, autoantibodies, which penetrate from the dermis, might have better access to the lower part of the epithelia.
In pregnant women with pemphigus, autoantibodies cross the placenta and bind to the fetal epidermis. However, neonates develop blisters if the mother has pemphigus vulgaris, but very rarely if she has pemphigus foliaceus. This confusing observation is also explained by the desmoglein compensation theory . The distribution of Dsg3 within neonatal epidermis is unlike that in adult epidermis; it is found on the surface of keratinocytes throughout the epidermis, which is similar to its distribution in mucous membranes (remember, neonatal skin is bathed in amniotic fluid). Therefore, pemphigus foliaceus sera containing only anti-Dsg1 IgG cannot efficiently induce blisters in neonatal skin.
As an extension of this compensation theory, exfoliative toxins, which are produced by Staphylococcus aureus and lead to bullous impetigo as well as staphylococcal scalded skin syndrome, specifically cleave Dsg1 . Inactivation of Dsg1 by this toxin induces superficial blisters in the epidermis that are clinically and histologically similar to those seen in pemphigus foliaceus.
In pemphigus, the disruption of cell–cell adhesion is currently thought to be mediated via the combined effects of direct inhibition by antibodies plus subsequent signal transduction induced by antibody binding . The direct inhibition is mediated by steric hindrance, i.e. the binding of autoantibodies to desmogleins spatially interferes with the adhesive interaction of desmogleins between cells. This proposed pathogenesis is supported by the following observations: (1) in pemphigus vulgaris and foliaceus, dominant epitopes are localized to the functionally important N-terminal regions of desmogleins ; and (2) pathogenic anti-Dsg3 mouse or human monoclonal antibody, but not non-pathogenic monoclonal antibodies, recognizes the N-terminal adhesive surface of Dsg3 . The phenotype of the Dsg3 null mouse (whose Dsg3 gene is genetically deleted) closely resembles that of human pemphigus vulgaris patients . The role of signal transduction induced by antibody binding is supported by the in vitro observation that IgG from pemphigus vulgaris sera (when added to the media of cultured keratinocytes) causes a transient increase in intracellular calcium and/or inositol 1,4,5-triphosphate, activation of protein kinase C, or phosphorylation of Dsg3 . Furthermore, with desmosomal disassembly, Dsg3 is internalized or endocytosed from the cell surface in association with keratin retraction .
Humoral and Cellular Autoimmunity in Paraneoplastic Pemphigus
Patients with paraneoplastic pemphigus develop characteristic IgG autoantibodies against multiple antigens , including Dsg3 and/or Dsg1 , multiple members of the plakin family (plectin, epiplakin, desmoplakins I and II, bullous pemphigoid antigen 1, envoplakin, and periplakin), and the protease inhibitor alpha-2-macroglobulin-like-1 (see Table 29.2 ). Anti-desmoglein antibodies play a role in inducing the loss of cell adhesion of keratinocytes and initiate blister formation, while the pathophysiologic relevance of the anti-plakin autoantibodies is unclear, in that plakin molecules are intracellular and IgG cannot penetrate cell membranes. In addition to humoral autoimmunity, cell-mediated cytotoxicity is involved in the pathogenesis of paraneoplastic pemphigus, in which more severe and refractory oral erosions and stomatitis as well as more polymorphic skin eruptions are seen, in comparison with classic forms of pemphigus. It was recently demonstrated in mice that Dsg3-specific T cells not only help B cells produce anti-Dsg3 IgG (which causes acantholysis), but also directly infiltrate into the epidermis and induce an interface dermatitis . Clarification of the exact roles of autoimmune T cells should provide valuable insights into the pathophysiology of paraneoplastic pemphigus.
Immunologic Mechanism of Pathogenic Autoantibody Production in Pemphigus
In contrast to the significant progress since the late 1980s in understanding the pathophysiologic mechanisms of blister formation in pemphigus, it is still unclear why patients with pemphigus begin to produce the pathogenic autoantibodies.
Pemphigus autoantibodies are composed of IgG isotypes, which may be produced after isotype switching, and they have a high affinity towards the antigen, which may be a result of affinity maturation of the antibodies. In addition, pemphigus sera recognize several distinct epitopes on desmogleins , and the presence of autoantibodies is associated with specific HLA class II alleles, including DRB1*0402, DRB1*1401 and DQB1*0302 in Caucasians and DRB1*14 and DQB1*0503 in Japanese . All of these features suggest that autoantibody production in pemphigus is T cell-dependent. More recently, T cells reactive against Dsg3 were shown to be present in peripheral blood from patients with pemphigus vulgaris as well as healthy individuals . Certain peptides from Dsg3, predicted to fit into the DRB1*0402 pocket, were able to stimulate T cells from the pemphigus patients.
Another advance that will allow the study of T cells and B cells is the development of an active disease mouse model for pemphigus vulgaris . This model is valuable not only for dissecting the cellular and molecular mechanisms involved in antibody production but also for developing novel therapeutic strategies.
Clinical Features
Pemphigus Vulgaris
Essentially all patients with pemphigus vulgaris develop painful erosions of the oral mucosa. More than half of the patients also develop flaccid blisters and widespread cutaneous erosions. Pemphigus vulgaris is therefore divided into two subgroups: (1) the mucosal-dominant type with mucosal erosions but minimal skin involvement; and (2) the mucocutaneous type with extensive skin blisters and erosions in addition to mucosal involvement (see Fig. 29.4 ).
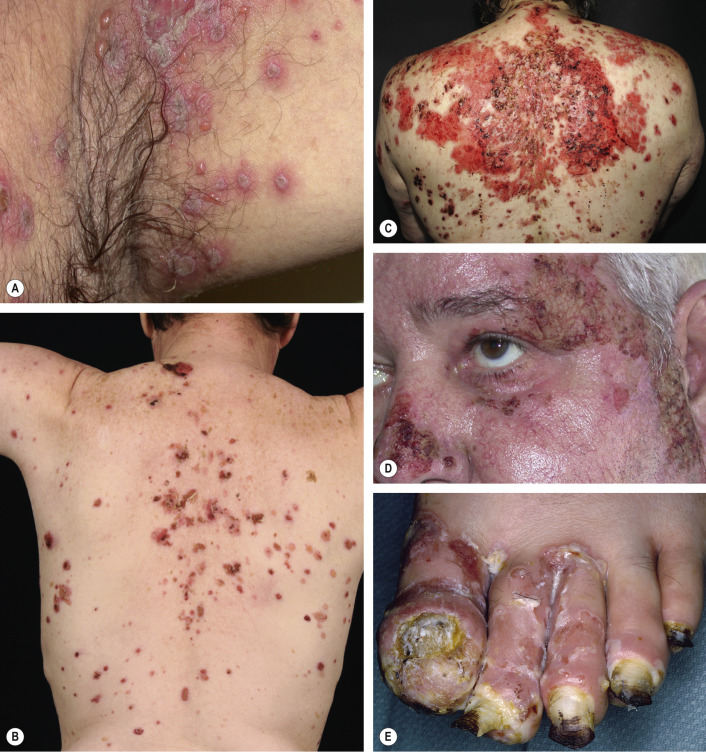
Mucous membrane lesions usually present as painful erosions ( Fig. 29.5 ). Intact blisters are rare, probably because they are fragile and break easily. Although scattered or extensive erosions may be seen anywhere in the oral cavity, the most common sites are the buccal and palatine mucosa. The erosions are of different sizes with an irregular and ill-defined border, which, when extensive or painful, may result in decreased oral intake of food or liquids. The diagnosis of pemphigus vulgaris tends to be delayed in patients presenting with only oral involvement, as compared to patients with skin lesions.
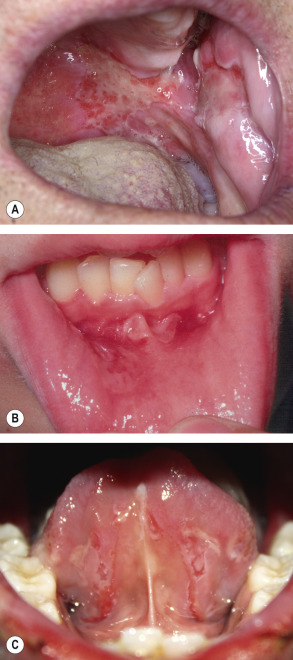
The lesions may extend out onto the vermilion lip and lead to thick, fissured hemorrhagic crusts. Involvement of the throat produces hoarseness and difficulty in swallowing. The esophagus also may be involved and sloughing of its entire lining in the form of a cast has been reported. The conjunctivae, nasal mucosa, vagina, labia, penis and anus can develop lesions as well. Cytology of vaginal cells may be misread as a malignancy when vaginal lesions are present.
The primary skin lesions of pemphigus vulgaris are flaccid, thin-walled, easily ruptured blisters ( Fig. 29.6 ). They can appear anywhere on the skin surface and arise on either normal-appearing skin or erythematous bases. The fluid within the bullae is initially clear but may become hemorrhagic, turbid, or even seropurulent. The blisters are fragile and soon rupture to form painful erosions that ooze and bleed easily. These erosions often attain a large size and can become generalized. The erosions soon become partially covered with crusts that have little or no tendency to heal. Those lesions that do heal often leave hyperpigmented patches with no scarring. Associated pruritus is uncommon. Table 29.3 outlines more unusual clinical presentations of pemphigus vulgaris.