Abstract
Bullous pemphigoid (BP), mucous membrane pemphigoid (MMP), and epidermolysis bullosa acquisita (EBA) are autoimmune blistering diseases of the skin and mucosae that are associated with autoantibodies directed against components of the dermal–epidermal or epithelial–stromal junction of stratified epithelia. These diseases exhibit clinical overlap. BP is often associated with generalized excoriated, eczematous, urticarial and/or bullous lesions, while MMP is characterized by blistering and scarring primarily of the external mucosal surfaces. Finally, EBA presents as either a mechanobullous disorder or an inflammatory BP-like disease, or, more rarely, as MMP. Their precise diagnosis relies on direct immunofluorescence microscopy as well as on the characterization of the specificity of the patient’s autoantibodies. Because of the significant morbidity and specific disease sequelae, proper evaluation and patient management are essential. Topical and systemic corticosteroids represent the mainstay of therapy; immunosuppressive medications are commonly used as steroid-sparing agents and novel biological therapies, particularly rituximab, may be employed.
Keywords
bullous pemphigoid, mucous membrane pemphigoid, epidermolysis bullosa acquisita, autoimmune bullous disease, immunofluorescence microscopy, cicatricial pemphigoid, ELISA
Bullous Pemphigoid
▪ Pemphigoid
- ▪
Bullous pemphigoid (BP) is the most common autoimmune subepidermal blistering disease, and its onset is often after 60 years of age
- ▪
It is usually a chronic disease, with spontaneous exacerbations and remissions, which may be accompanied by significant morbidity
- ▪
BP is associated with tissue-bound and circulating autoantibodies directed against BP antigen 180 (BP180, BPAG2 or type XVII collagen) and BP antigen 230 (BP230 or BPAG1e), components of junctional adhesion complexes called hemidesmosomes that promote dermal–epidermal cohesion
- ▪
The spectrum of clinical presentations is extremely broad. Characteristically, BP is an intensely pruritic eruption with widespread blister formation. In early stages, or in atypical variants of the disease, only eczematous or urticarial lesions (either localized or generalized) are present or just excoriations due to intense pruritus. Occasionally, patients present with just pruritus
- ▪
Diagnosis relies on immunopathologic examinations, particularly direct and indirect immunofluorescence microscopy as well as ELISA for anti-BP180/BP230 autoantibodies
Introduction
Bullous pemphigoid (BP) is the most common autoimmune subepidermal blistering disease. It typically presents in older adults as a generalized pruritic bullous eruption and is potentially associated with significant morbidity. The clinical presentation can be quite polymorphic, particularly during the early stages of the disease or in atypical variants, in which full-blown bullous lesions may be absent. In these cases, establishing the diagnosis of BP requires a high degree of suspicion. The antigens targeted by patients’ autoantibodies represent two components of hemidesmosomes, the junctional adhesion complexes found in skin and mucosae (see Fig. 28.3A ).
History
During the eighteenth century, the term “pemphigus” was often used to describe any type of bullous eruption. It was not until 1953 that Lever, on the basis of specific clinical and histologic features, recognized BP as a disorder distinct from the various types of “true” pemphigus .
A decade later, Jordon, Beutner and colleagues demonstrated that BP patients had tissue-bound and circulating autoantibodies directed against the cutaneous basement membrane zone (BMZ), an observation that suggested that subepidermal dysadhesion was due to autoantibodies directed against structural components of the skin that promoted dermal–epidermal adhesion . Further milestones in our understanding of BP included the immunochemical characterization of targeted proteins, the cloning of their genes, and the development of animal models of the disease .
Epidemiology
BP is typically a disease of older adults, with an onset after 60 years of age. The annual incidence has been estimated to be at least 6–13 new cases per million population; however, recent studies suggest a three-fold higher incidence, most likely due to improved survival amongst the elderly as well as the recognition of non-bullous variants . The relative risk for patients over 90 years of age appears to be about 300-fold higher than for those 60 years of age or younger, with an apparent higher predominance in men than in women. The disease also occurs in children, but rarely. Certain HLA class II alleles are more prevalent in patients with BP than in the general population . In Caucasians, a significant association with the allele DQB1*0301 has been found, while an increased frequency of the alleles DRB1*04, DRB1*1101, and DQB1*0302 has been observed in Japanese patients.
Pathogenesis
BP is an example of an immune-mediated disease that is associated with a humoral and cellular response directed against two well-characterized self-antigens: BP antigen 180 (BP180, also known as BPAG2 or type XVII collagen) and BP antigen 230 (BP230, also referred to as the epithelial isoform of BPAG1 [BPAG1e]) ( Table 30.1 ) . While the former is a transmembrane protein with a large collagenous extracellular domain, the latter is a cytoplasmic protein belonging to the plakin family (see Ch. 28 ). These two antigens are components of the hemidesmosomes, which are adhesion complexes promoting epithelial–stromal adhesion in stratified and other complex epithelia. Immunoelectron microscopy studies utilizing gold labeling have demonstrated that in vivo -deposited IgG antibodies are localized to the hemidesmosomal plaque and to the outside of the basal cell plasma membrane beneath the hemidesmosome, in a distribution corresponding to the locations of BP230 and of BP180, respectively.
| MAJOR AUTOANTIGENS OF SUBEPIDERMAL AUTOIMMUNE-MEDIATED BLISTERING DISEASES | |||
|---|---|---|---|
| Disease | Target antigen(s) | Mol. wt. (kDa) | Morphologic structures |
| Bullous pemphigoid (BP) | BP180/BPAG2/collagen XVII | 180 | Hemidesmosomal plaque/anchoring filaments |
| BP230/BPAG1e | 230 | Hemidesmosomal plaque | |
| Gestational pemphigoid | BP180/BPAG2/collagen XVII | 180 | Hemidesmosomal plaque/anchoring filaments |
| BP230/BPAG1e | 230 | Hemidesmosomal plaque | |
| Mucous membrane (cicatricial) pemphigoid | BP180/BPAG2/collagen XVII | 180 | Hemidesmosomal plaque/anchoring filaments |
| BP230/BPAG1e † | 230 | Hemidesmosomal plaque | |
| Laminin 332 (laminin 5; α 3 β 3 γ 2 ; epiligrin) | 165, 140, 105 | Anchoring filaments | |
| Laminin 311 (laminin 6; α 3 β 1 γ 1 ) ‡ | 165, 220, 200 | Anchoring filaments/extracellular matrix | |
| Integrin β 4 subunit § | 200 | Hemidesmosomal plaque/anchoring filaments | |
| Linear IgA bullous dermatosis (LABD) | LAD antigen ¶ | 97/120 | Anchoring filaments |
| BP180/BPAG2/collagen XVII | 180 | Hemidesmosomal plaque/anchoring filaments | |
| BP230/BPAG1e † | 230 | Hemidesmosomal plaque | |
| Type VII collagen † | 290/145 | Anchoring fibrils | |
| Epidermolysis bullosa acquisita | Type VII collagen † | 290/145 | Anchoring fibrils |
| Anti-laminin γ1 pemphigoid (previously known as anti-p200 pemphigoid) | Laminin gamma-1 chain | 200 | Extracellular matrix |
| Bullous systemic lupus erythematosus | Type VII collagen † | 290/145 | Anchoring fibrils |
† Detectable in a subset of patients.
‡ Binding to laminin 311 depends on the presence of cross-reactive autoantibodies directed against the α-chain of laminin 332 (laminin 5).
§ Reactivity with the cytoplasmic domain of the β 4 subunit of the α 6 β 4 integrin described in a subset of patients with ocular cicatricial pemphigoid.
¶ It constitutes the most characteristic serologic marker for LABD. The 120 kDa LAD antigen corresponds to the cleaved, shed extracellular domain of BP180/BPAG2. The 97 kDa protein results from its further proteolytic degradation (see Fig. 31.9 ).
In vitro studies and in vivo animal models have provided strong evidence for the pathogenic role of autoantibodies in BP. Furthermore, in gestational pemphigoid, a disease closely related to BP, the transplacental transfer of autoantibodies to BP180 from the mother to the neonate can result in a transient bullous eruption (see Ch. 34 ).
Humoral and cellular responses
Almost all patients with BP have circulating IgG autoantibodies that bind to BP180. More specifically, it is the non-collagenous NC16A domain, a region of BP180 located extracellularly but close to the transmembrane domain, that constitutes the immunodominant region (see Fig. 31.9 ) . However, additional antigenic sites exist within both the extracellular and intracellular domains of BP180, and they are recognized by up to 70% of BP sera . Patients with BP also exhibit significant autoreactivity to BP230 , and BP230-reactive autoantibodies bind predominantly to the C-terminal region of this autoantigen . The presence of several antigenic sites throughout BP180 and BP230 most likely results from the phenomenon known as “epitope spreading” (see below) . This phenomenon may also account for the finding that patients’ sera rarely contain autoantibodies targeting additional components of the BMZ.
Patients with BP develop an autoreactive T-cell response to BP180 and BP230, and this is probably crucial for stimulating B cells to produce autoantibodies . The responsiveness of anti-BP180 autoreactive T cells is restricted by certain HLA class II alleles (e.g. HLA-DQB1*0301), which are prevalent in BP patients. These T lymphocytes, whose major relevant epitopes seem to be harbored within the NC16 domain, have a CD4 + phenotype and produce both Th1 (e.g. interferon-γ) and Th2 cytokines (e.g. interleukin [IL]-4, IL-5 and IL-13; see Ch. 4 ) . Th2, as well as more recently identified Th17, cytokines may be particularly relevant to the pathophysiology of BP ; they predominate within lesional tissue and in patients’ sera. Also, the IgG4 subclass, the secretion of which is regulated by Th2 cytokines, is one of the major isotypes of anti-BP180 autoantibodies.
Upon binding of autoantibodies to their target antigens, subepidermal blister formation results from a cascade of events in which Fc receptor-mediated mechanisms, via BP180–anti-BP180 antibody complexes, are critical . The latter involve complement activation and recruitment of inflammatory cells, with liberation of their proteases including matrix metalloproteinase-9, neutrophil elastase, and mast cell proteases . These proteinases proteolytically degrade various extracellular matrix proteins as well as BP180. Infiltrating innate immune cells (macrophages, neutrophils, mast cells, and eosinophils) contribute to tissue damage via the release of proinflammatory cytokines, e.g. IL-4, -5, -8, -17, and eotaxin, which further enhance the inflammatory response . Autoantibodies to BP180 can also amplify the inflammatory response by directly stimulating keratinocytes to express inflammatory cytokines ( Fig. 30.1 ) . Finally, IgG autoantibodies reduce hemidesmosomal BP180 content and thereby contribute in a complement-independent manner to weakening of dermal–epidermal cohesion .
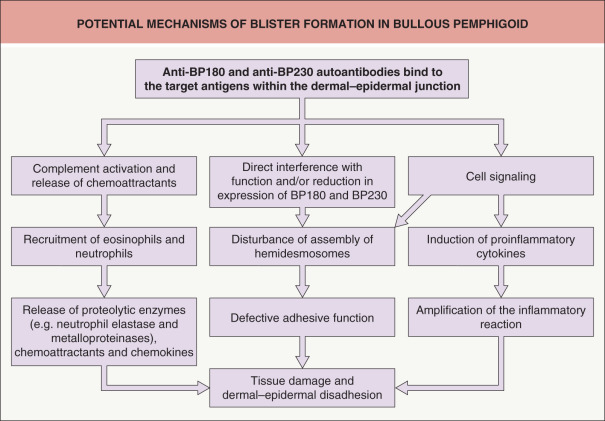
Various animal models have provided strong evidence that autoantibodies against BP180 are pathogenic. When passively transferred to neonatal mice (in which BP180 had been fully or partially humanized by genetic engineering), human autoantibodies against the NC16A domain were able to induce a blistering disorder that reproduced all the key features of BP . In a different murine model, anti-BP230 antibodies induced an inflammatory reaction and subepidermal blistering . Nonetheless, the currently accepted pathomechanism is that antibodies against the ectodomain of BP180 are critical for disease initiation, while the development of antibodies against BP230 represents a secondary event that contributes to tissue damage .
Clinical Features
Non-bullous pemphigoid
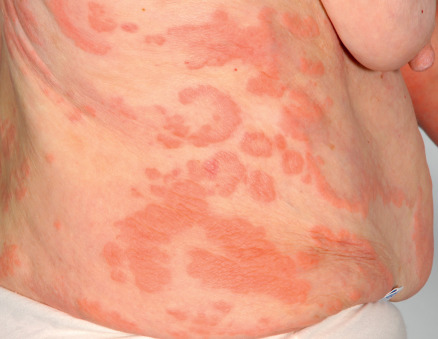
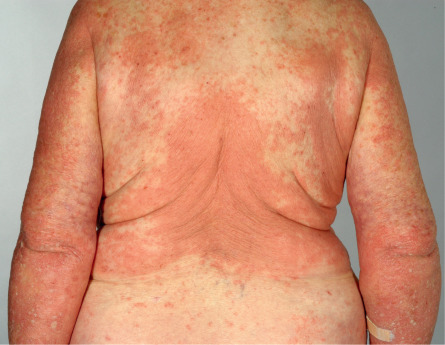
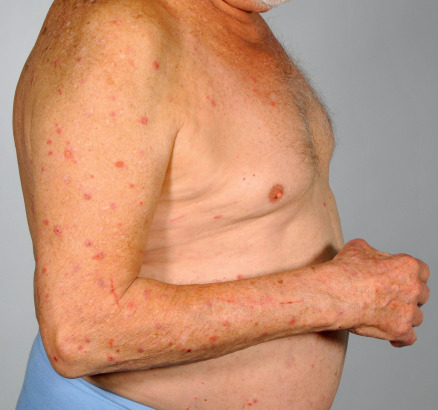
The cutaneous manifestations of BP can be extremely polymorphic ( Figs 30.2–30.6 ) . In the prodromal, non-bullous phase of the disease, signs and symptoms are frequently nonspecific, with mild to severe intractable pruritus alone or in association with excoriated, eczematous, papular and/or urticarial lesions that may persist for several weeks or months (see Figs 30.3–30.5 ). Importantly, these nonspecific skin findings may remain as the only signs of the disease. More recent studies have noted that at least 20% of patients have neither obvious blisters nor erosions due to ruptured bullae at the time of diagnosis .
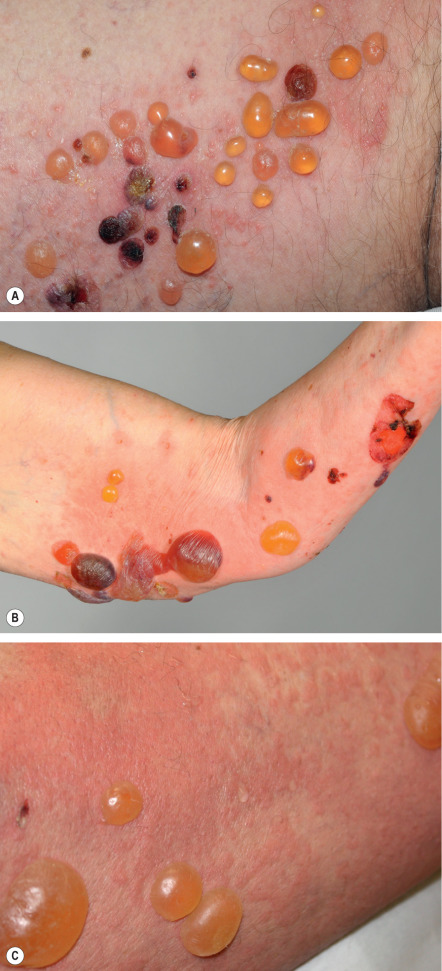
Bullous phase
The bullous stage of BP is characterized by the development of vesicles and bullae on apparently normal or erythematous skin, together with urticarial and infiltrated papules and plaques that occasionally assume an annular or figurate pattern. The blisters are tense, up to 1–4 cm in diameter, contain a clear fluid, and may persist for several days, leaving eroded and crusted areas (see Fig. 30.2 ). Occasionally, the blister fluid becomes blood-tinged. The lesions frequently have a symmetrical distribution pattern, and they predominate on the flexural aspects of the extremities and the lower trunk, including the abdomen. Within intertriginous zones, vegetating plaques may be observed (pemphigoid vegetans; see Fig. 30.6C ). The extent and degree of disease activity can be assessed by employing the BP disease area index (BPDAI) or a daily blister count.
Residual postinflammatory changes include hyper- and hypopigmentation and, only occasionally, milia. Involvement of the oral cavity is observed in 10–30% of patients. The mucosae of the eyes, nose, pharynx, esophagus, and anogenital region are more rarely affected. In ~50% of patients, a peripheral blood eosinophilia is noted.
Clinical variants
Several clinical variants of BP have been described and are outlined in Table 30.2 . Pemphigoid gestationis (or gestational pemphigoid ) is also a variant of BP, which typically occurs during pregnancy (see Ch. 27 ).
| UNUSUAL CLINICAL VARIANTS OF BULLOUS PEMPHIGOID | |
|---|---|
| |
| |
* Also referred to as “stump“ pemphigoid.
† Radiotherapy can also provoke generalized form of pemphigoid.
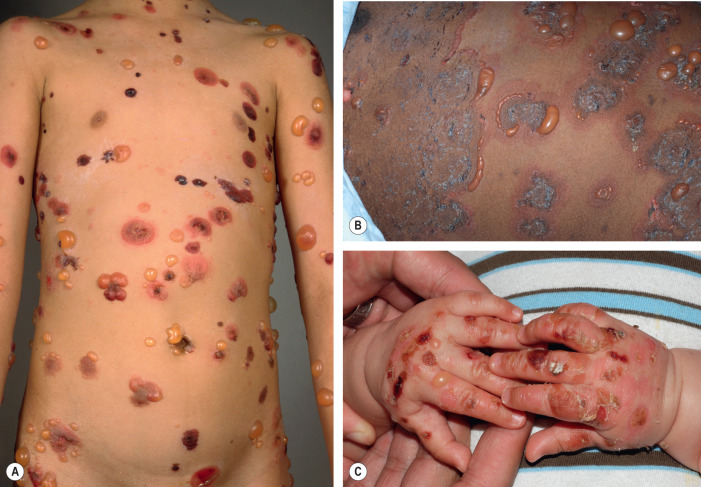
While the individual lesions of BP in infants and in older children ( infantile and childhood BP ) are similar to those observed in older adults, the sites of involvement can differ ( Fig. 30.7 ). In infants, bullae often first appear acrally and then can generalize to other sites, including the face. Involvement of the genital region, e.g. vulvar childhood pemphigoid , as well as other mucosal sites has been observed in children .
Associated Diseases
The association of internal malignancies with BP is probably related primarily to the older age of the patient. In case–control studies, the trend towards an increased risk of malignancy was marginal , and a recent English national record linkage study found no increased risk for concurrent or subsequent malignancies in BP patients . Nonetheless, patients with BP should be up-to-date with age-related cancer screening tests recommended for the general population.
Rarely, BP has been described in patients with inflammatory bowel disease or other autoimmune disorders such as Hashimoto thyroiditis, rheumatoid arthritis, dermatomyositis, and lupus erythematosus (LE). It is thought that these associations are not fortuitous but reflect a genetically determined susceptibility to develop autoimmune diseases . However, a case–control study did not find any increased risk for autoimmune disorders in those with BP .
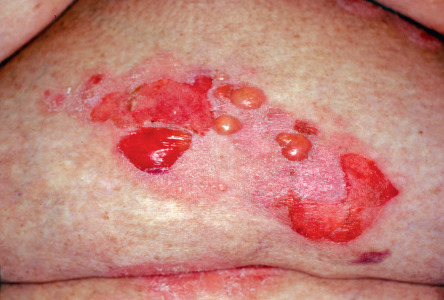
In some patients, BP appears to be triggered by trauma, burns, radiotherapy or UV irradiation (including PUVA). BP has also been found in association with certain dermatoses, such as psoriasis and lichen planus, and the bullae may be localized to the psoriatic plaques ( Fig. 30.8 ). It has been speculated that a chronic inflammatory process at the dermal–epidermal junction results in the exposure of antigens to autoreactive T lymphocytes, leading to a secondary immune response (epitope spreading phenomenon).
Finally, BP is significantly associated with neurological disorders, such as Parkinson disease, dementia, psychiatric disorders (unipolar and bipolar disorders), and stroke. A strong association with multiple sclerosis was also observed in a population-based study . It should be noted that neuronal variants of BP230 are expressed in the central and peripheral nervous systems .
Drug-induced bullous pemphigoid
In certain patients, systemic medications can lead to the development of BP . The implicated drugs are numerous, including diuretics (e.g. furosemide, spironolactone), NSAIDs (e.g. ibuprofen, topical diclofenac), antibiotics (e.g. amoxicillin, ciprofloxacin), ACE inhibitors, TNF inhibitors, potassium iodide, vaccines, and more recently, dipeptidyl peptidase-4 inhibitors (e.g. vildagliptin) and checkpoint inhibitors (e.g. pembrolizumab). Reproduction of BP lesions following drug rechallenge has been observed with some medications (e.g. furosemide), but for others, the association is based on less evidence. Case–control studies assessing the drugs used on a long-term basis prior to onset of the disease found that two classes of drugs, diuretics and neuroleptics, were used more frequently by BP patients than by control subjects. With regard to diuretics, the risk was variably linked to aldosterone antagonists and loop diuretics . Hence, in all patients, a careful drug history is mandatory to exclude the triggering effect of a drug, since its prompt discontinuation may result in improvement.
It is likely that the drugs act as triggers in patients with an underlying genetic susceptibility by either modifying the immune response or altering the antigenic properties of the epidermal basement membrane.
Diagnosis
The diagnosis of BP is based upon the typical clinical presentation, compatible histologic features, and, most importantly, positive direct immunofluorescence (DIF) microscopy studies (see Fig. 29.17 ) . Further support for the diagnosis of BP requires the detection of specific circulating IgG autoantibodies (anti-BP180, anti-BP230) via either indirect immunofluorescence (IIF) microscopy or ELISA. In the vast majority of patients, these tests allow for the correct classification of patients. However, in a minority of patients (~10%) in whom both IIF microscopy and ELISA are negative, additional immunopathological studies, e.g. n-serration versus u-serration pattern analysis (see below), can be employed to demonstrate an autoantibody response to BP180 and/or BP230 and thereby exclude other autoimmune bullous diseases (see Table 30.1 ) .
Light microscopy
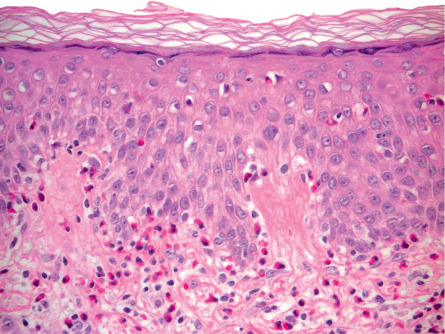
In the non-bullous phase or in atypical variants of BP, routine histology may provide less specific information, since only eosinophilic spongiosis and/or dermal infiltrates of eosinophils may be seen ( Fig. 30.9 ). In biopsy specimens of an early bulla, a subepidermal blister accompanied by a dermal inflammatory infiltrate composed of eosinophils and mononuclear cells is typically observed. The infiltrate favors the uppermost dermis, and the cavity of the bulla contains a net of fibrin with a variable inflammatory infiltrate ( Fig. 30.10 ). Electron microscopy studies have shown that subepidermal blister formation occurs at the level of the lamina lucida.
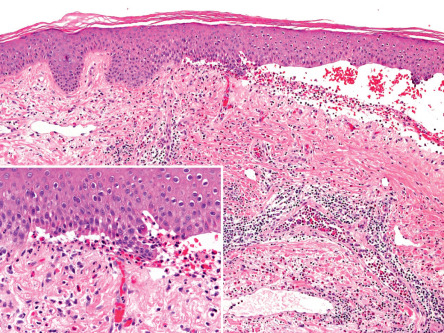
Direct immunofluorescence (DIF) microscopy
In almost all patients, DIF microscopy of perilesional, uninvolved skin will characteristically demonstrate the presence of fine, linear, continuous deposits of IgG and/or C3 (and, more rarely, other Ig classes) along the epidermal basement membrane (see Ch. 27 ). IgG4 and IgG1 are the predominant IgG subclasses. Two additional studies are helpful in distinguishing BP from other autoimmune blistering disorders: (1) close analysis of the linear fluorescence pattern at the BMZ in order to determine if it is n-serrated (BP and linear IgA bullous dermatosis) versus u-serrated (epidermolysis bullosa acquisita) ; and (2) the salt-split skin assay in which perilesional skin is examined after treatment with 1 M NaCl (see Fig. 28.7 ). In BP, immune deposits are found in the epidermal side (roof) or in both the epidermal and dermal sides of the split. Although not routinely available, the computer-aided fluorescence overlay antigen mapping (FOAM) technique allows one to determine more precisely the localization of deposited immunoreactants .
Indirect immunofluorescence (IIF) microscopy
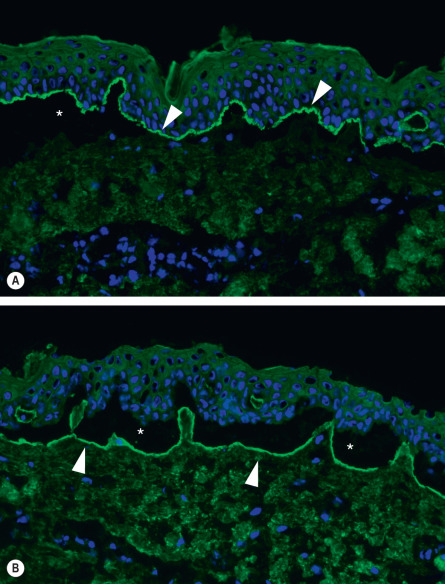
For IIF studies in subepidermal blistering diseases, salt-split normal human skin is the substrate of choice, rather than intact normal human skin or monkey esophagus (see Table 29.5 ). Circulating anti-basement membrane autoantibodies of the IgG class, and, less frequently, of the IgA and IgE classes, are detectable in 60–80% of patients . These autoantibodies typically bind to the epidermal side or, less frequently, to both the epidermal and dermal sides of saline-separated normal human skin ( Fig. 30.11 ) . A BIOCHIP IIF technique has also been introduced in which the substrates are recombinant tetrameric BP180-NC16A and transfected cells expressing BP230 .
ELISA
An increasing number of autoantibodies associated with autoimmune bullous diseases can be detected via ELISA (see Table 29.6 ). Target antigens include the NC16A domain of BP180 and the C-terminus (+/− the N-terminus) of BP230 . In the case of BP, these tests have been found to be fairly specific (≥90%); occasionally, low-titer, false-positive results are observed in healthy subjects and elderly patients with pruritic cutaneous eruptions . When performed in unselected BP patients, the overall sensitivity of the BP180-NC16A ELISA is comparable to that of IIF (with salt-split skin as a substrate) . Combining the ELISA for BP230 with the BP180-NC16A ELISA increases the overall sensitivity by ~10% so the former is only recommended in the setting of a negative BP180 ELISA . In contrast to immunoblotting, ELISA antigens are tested under native conditions, and, as a result, binding activity against conformational antigens is not lost.
Other immunopathological studies
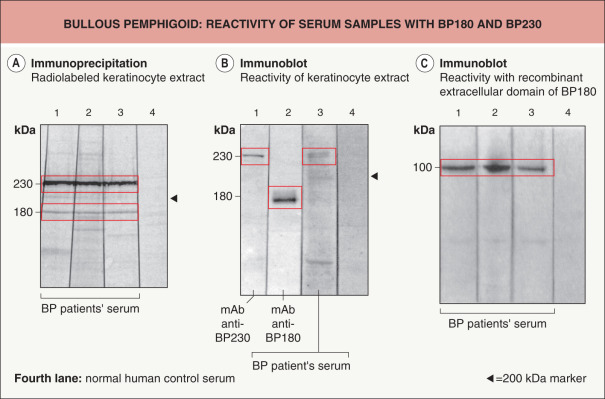
In immunoblot and immunoprecipitation studies of keratinocyte extracts, 60–100% of patients’ sera contain IgG autoantibodies that bind to BP180 and BP230, respectively ( Fig. 30.12A,B ) . Patients’ sera also frequently contain specific IgA and IgE autoantibodies. In general, recombinant forms of BP180 and BP230 expressed in prokaryotic or eukaryotic systems are used for the detection of autoantibodies ( Fig. 30.12C ) .
Differential Diagnosis
Because the clinical findings in the non-bullous phase of BP may be nonspecific, they can resemble a variety of dermatoses, including drug reactions, contact dermatitis, prurigo (simplex and nodularis), urticarial dermatoses, arthropod reactions, and scabies . These disorders are usually distinguished on the basis of the clinical history and setting, pathologic features, and the negative findings with IF microscopy. The presence of bullae raises the possibility of bullous arthropod bites, allergic contact dermatitis, Stevens–Johnson syndrome, bullous drug eruptions (see Ch. 33 ), dyshidrotic eczema, pseudoporphyria or porphyria cutanea tarda. In children, bullous impetigo, inherited epidermolysis bullosa, and bullous mastocytosis must also be considered.
The pemphigus group, paraneoplastic pemphigus, and dermatitis herpetiformis can be differentiated on the basis of distinctive immunopathologic findings and clinical context. A study found that, in patients with a subepidermal blistering disorder associated with linear deposits of IgG or C3 along the epidermal basement membrane, the presence of the following four clinical criteria strongly indicated a diagnosis of BP: (1) absence of skin atrophy; (2) absence of mucosal involvement; (3) absence of head and neck involvement; and (4) age greater than 70 years . Nevertheless, the distinction of BP from the following autoimmune subepidermal blistering disorders can sometimes prove challenging (see Table 30.1 ).
- •
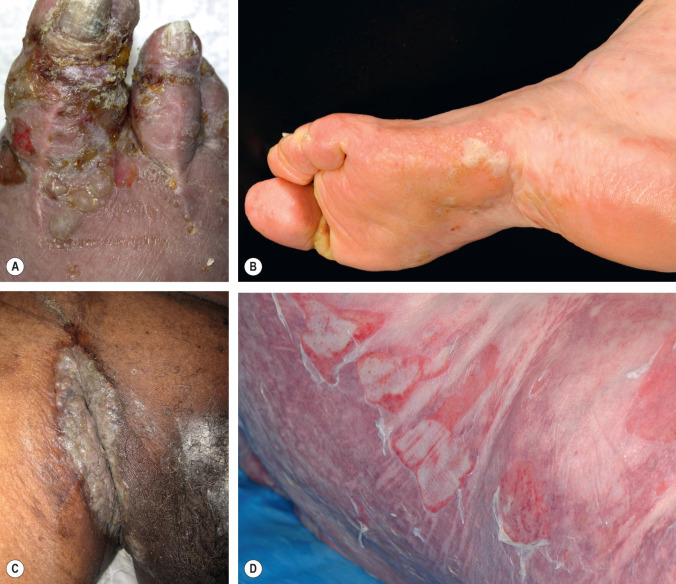
Epidermolysis bullosa acquisita (EBA) has a wide spectrum of clinical presentations (see below). While the classic “non-inflammatory” form of EBA is sufficiently distinctive, the “inflammatory” form closely mimics BP. As with BP, mucosal involvement may be present.
- •
Linear IgA bullous dermatosis (LABD) represents a group of subepidermal blistering diseases rather than a single entity (see Ch. 31 ). While features of LABD are polymorphic in adults, during childhood the condition is frequently associated with annular and polycyclic lesions as well as involvement of the genital and perioral region. However, identical features are also observed in childhood BP (see Fig. 30.7B ).
- •
Mucous membrane (cicatricial) pemphigoid is a heterogeneous group of diseases which have in common the predominant involvement of the mucosae, a chronic course, and a scarring tendency (see below). Skin lesions are only found in up to 25–30% of patients, and they generally involve the head and upper trunk. In patients with both oral and cutaneous lesions, the differentiation of mucous membrane pemphigoid from BP is difficult, and classification relies on the presence of an obvious scarring tendency of involved mucosal sites and limited skin involvement, and, occasionally, on the results of immunologic assays (see Table 30.1 ).
- •
Pemphigoid incipiens . A difficult issue is how to categorize the group of elderly patients with generalized pruritus (with or without skin lesions), in whom circulating autoantibodies to the epidermal BMZ and reactivity with BP180 and/or BP230 are found, but routine IF microscopy remains negative. Some of these patients, with initially negative DIF microscopy findings, eventually do develop BP and could be thought of as having pemphigoid incipiens .
- •
Anti-laminin γ1 pemphigoid (previously known as anti-p200 pemphigoid) . A small group of patients have been described with features identical to those observed in BP, i.e. vesicles and tense blisters as well as eczematous and urticarial papules and plaques. Occasionally, grouped papulovesicles with a dermatitis herpetiformis-like presentation are also present, and there may be mucosal involvement. Neutrophils, rather than eosinophils, are usually the predominant cells within the inflammatory infiltrate. These patients have circulating autoantibodies that specifically bind the dermal side of salt-split human skin. The targeted 200 kDa BMZ protein is the gamma 1 chain of laminin (see Table 30.1 ) .
Prognosis
BP is a chronic disease characterized by spontaneous exacerbations. Approximately 30% of BP patients have a relapse during their first year of treatment, with extensive disease and associated dementia as independent risk factors for relapse. Furthermore, after cessation of therapy, ~50% of patients experience a relapse, most often within the first 3 months .
Mortality is considerable among elderly patients. The estimated death rate during the first year varies between 10% and 40%, depending on the series . Age and a Karnofsky score that is <40 (range, 0–100) have been shown to significantly affect prognosis . It is likely that comorbidities and practice patterns (use of systemic corticosteroids and/or immunosuppressive drugs) also influence overall morbidity and mortality. Quality of life is obviously an issue for many patients given the associated symptoms.
Monitoring
The practicality of using the results of quantitative serologic tests, such as the BP180 ELISA, as a means of guiding treatment remains to be established. That said, serum levels of IgG autoantibodies to BP180 did correlate with disease severity in several ELISA-based studies . Furthermore, determination of anti-BP180 IgG antibody levels by ELISA at days 0, 60, and 150 appeared to help predict disease relapse – a small (<20%) decrease in serum autoantibody levels between days 0 and 60 was associated with relapse during the first year of therapy . Finally, a high BP180-NC16A ELISA score (>27 U/ml) and, to a lesser degree, positive DIF findings at cessation of therapy are both good indicators of future relapse of BP.
Treatment
The choice of treatment depends on the severity of the disease and comorbidities ( Tables 30.3 & 30.4 ) . For extensive disease, defined by some authors as either >10 new blisters/day or inflammatory lesions involving a large body surface area, a regimen of oral prednisone at a dose of 0.5–1 mg/kg/day is often recommended. This dose usually controls the disease within 1 or 2 weeks and is then progressively tapered over a period of 6–9 months, or occasionally longer. Some authors begin the prednisone taper once there have been no new lesions or pruritus for at least 2 weeks . However, the use of systemic corticosteroids, especially in the elderly, is associated with significant side effects (see Ch. 125 ) .
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


