Abstract
Palmoplantar keratoderma (PPK) represents a heterogeneous group of hereditary and acquired disorders of cornification characterized by hyperkeratosis of the skin on the palms and soles. Inherited PPK may occur as an isolated condition, a component of another genodermatosis (e.g. ichthyosis, epidermolysis bullosa, ectodermal dysplasia), or a part of a multisystem syndrome (e.g. with deafness or cardiomyopathy). Acquired forms can be drug-induced or paraneoplastic. PPKs have three major patterns of palmoplantar involvement: diffuse, focal (areata or striate), and punctate. Some PPKs demonstrate transgrediens, i.e. extension beyond volar skin. Patients may suffer from hyperhidrosis, maceration, blisters, fungal infections, malodor, constricting keratotic bands (pseudoainhum), and severe pain. A skin biopsy can sometimes provide clues to the specific diagnosis. The underlying molecular defect may involve epidermal proteases as well as proteins of the keratinocyte cytoskeleton, cornified envelope, desmosome, or gap junction.
Keywords
palmoplantar keratoderma (PPK), punctate PPK, focal PPK, diffuse PPK, hereditary PPK, syndromic PPK, acquired PPK, epidermolytic PPK, non-epidermolytic PPK, mutilating PPK, Vohwinkel syndrome, Huriez syndrome, Clouston syndrome, Olmsted syndrome, Papillon–Lefèvre syndrome, Naxos disease, Carvajal syndrome, pachyonychia congenita, spiny keratoderma, marginal papular keratoderma, acrokeratoelastoidosis, focal acral hyperkeratosis, keratoderma climactericum, aquagenic PPK
- ▪
Palmoplantar keratodermas can be inherited or acquired
- ▪
The three major patterns of involvement are diffuse, focal, and punctate
- ▪
The clinical presentation may be different on the hands and feet, e.g. diffuse on the soles and focal on the palms
- ▪
Additional distinguishing features include an erythematous border, transmigration to areas beyond the palmoplantar skin, and digital constrictions (pseudoainhum)
- ▪
Patients may also suffer from hyperhidrosis, maceration, blistering, fungal infections, malodor, and pain
- ▪
It is important to determine whether other features are present, e.g. nail dystrophy, hypotrichosis, oral lesions, hyperkeratosis of non-volar skin, impaired hearing, ocular findings, cardiomyopathy (in patients with woolly hair)
- ▪
Histologic examination sometimes shows specific findings, such as epidermolytic hyperkeratosis
Introduction
Palmoplantar keratodermas (PPKs) represent a heterogeneous group of hereditary and acquired disorders of cornification characterized by prominent hyperkeratosis of the skin on the palms and soles. Inherited PPK sometimes represents a component of a syndromic phenotype with extracutaneous manifestations such as cardiomyopathy or deafness. Likewise, acquired PPK can be drug-induced or associated with cancer. Determination of the specific subtype of PPK and recognition of associated features are therefore important ( Table 58.1 ).
| PRIMARY PALMOPLANTAR KERATODERMAS (PPKs) | |
| Disorder | Additional features |
| Hereditary PPKs | |
| Diffuse PPK, isolated | |
| Vörner–Unna–Thost | Epidermolytic |
| Nagashima, Kimonis | Non-epidermolytic |
| Bothnia | White/spongy upon water exposure; non-epidermolytic |
| “Greither” | Transgrediens & progrediens; variable pathology |
| Mal de Meleda (including Gamborg Nielsen) | Mutilating, frequent superinfections; non-epidermolytic |
| Diffuse PPK with associated features | |
| Hearing impairment: | |
| Vohwinkel syndrome | Mutilating, “honeycomb” PPK; starfish keratoses on knuckles |
| Bart–Pumphrey syndrome | Knuckle pads, leukonychia |
| PPK due to connexin 26 defect | “Honeycomb” PPK |
| Mitochondrial PPK | |
| High risk of acral SCC * : | |
| Huriez syndrome | Scleroatrophy |
| PPK with sex reversal | Female-to-male sex reversal |
| Hypotrichosis & nail dystrophy: | |
| Clouston syndrome | Pebbled/grid-like acral papules |
| Odonto-onycho-dermal dysplasia | Hypodontia, facial telangiectasias/erythema |
| Schöpf–Schulz–Passarge syndrome | Hidrocystomas, other adnexal neoplasms, hypodontia |
| Keratoderma–hypotrichosis–leukonychia totalis | Total leukonychia |
| Mutilating * : | |
| Olmsted syndrome | Periorificial/intertriginous keratotic plaques |
| Papillon–Lefèvre syndrome ** | Periodontitis, pyogenic infections, psoriasiform plaques |
| Loricrin keratoderma | Mild generalized ichthyosis, “honeycomb” PPK |
| KLICK | Keratosis linearis, ichthyosis congenita, sclerosing keratoderma |
| Vohwinkel syndrome (see above) | |
| Mal de Meleda (see above) | |
| Cardiomyopathy: | |
| Naxos disease *** | Woolly hair, arrhythmias in adolescence |
| Focal PPK, isolated | |
| Brünauer–Fuhs–Siemens, Wachters | Striate on palms, nummular on soles; see Table 58.6 for genetic subtypes |
| Focal PPK with associated features | |
| Pachyonychia congenita | Painful soles, dystrophic nails, oral leukokeratosis, steatocystomas/other cysts |
| Focal palmoplantar and gingival keratosis | Painful soles, gingival leukokeratosis; epidermolytic |
| Howel–Evans syndrome | Esophageal carcinoma |
| Richner–Hanhart syndrome (oculocutaneous tyrosinemia) | Dendritic keratitis, corneal ulcers, intellectual disability, painful keratosis |
| Carvajal syndrome *** | Striate on palms; woolly hair, dilated cardiomyopathy; occasionally skin fragility, nail dystrophy, and hypodontia |
| PPK and woolly hair | Striate on palms; woolly hair, hypotrichosis, leukonychia |
| Punctate PPK, isolated | |
| Buschke–Fischer–Brauer (type 1 punctate PPK) Spiny keratoderma (type 2 punctate PPK) Marginal papular keratoderma: acrokeratoelastoidosis, focal acral hyperkeratosis (type 3 punctate PPK) Punctate keratoses of the palmar creases Porokeratosis punctata palmaris et plantaris | |
| Punctate PPK with associated features | |
| Cole disease | Guttate hypopigmentation, calcinosis cutis |
| PLACK syndrome | P eeling skin, l eukonychia, c heilitis, k nuckle pads |
| Acquired PPKs | |
| Keratoderma climactericum Aquagenic PPK Pityriasis rubra pilaris Associated with hypothyroidism/myxedema Associated with cancer Associated with lymphedema Arsenical keratoses Drug-induced keratoderma (e.g. lithium, verapamil, venlafaxine, glucan, tegafur, capecitabine, imatinib, sorafenib, sunitinib) Induced by dioxin or halogenated herbicides Associated with dermatomyositis: pityriasis rubra pilaris-like (Wong) type, “mechanic’s hands” in antisynthetase syndrome | |
* Acral SCC can also develop in patients with mutilating PPK.
** Haim–Munk syndrome is an allelic disorder that also features arachnodactyly, atrophic nail changes, and acro-osteolysis.
*** The PPK in Naxos disease is occasionally focal/striate, overlapping with Carvajal syndrome.
Several classification systems for PPKs have been proposed, but none of these satisfactorily incorporates clinical presentation, pathology, and molecular pathogenesis. Current nomenclature is non-uniform and includes many eponyms . In addition, PPK may represent a component of other genodermatoses such as ichthyoses, erythrokeratodermas, epidermolysis bullosa, and ectodermal dysplasias ( Table 58.2 ).
| OTHER GENODERMATOSES THAT FEATURE PALMOPLANTAR KERATODERMA (PPK) |
| Erythrokeratodermas (see Ch. 57 ) |
| Other ichthyoses (see Ch. 57 ) |
|
| Other ectodermal dysplasias (see Chs 63 and 67 ) |
| Epidermolysis bullosa (EB; see Ch. 32 ) |
| Other |
|
* PPK is a major feature of the disorder.
** Include congenital ichthyosiform erythroderma, lamellar ichthyosis, and harlequin ichthyosis.
*** Features include nail dystrophy/loss, marginal PPK, hypodontia, enamel hypoplasia, oral hyperpigmentation, dysphagia, and deafness.
A simple working classification divides PPKs into three major types based on the clinical pattern of involvement :
- •
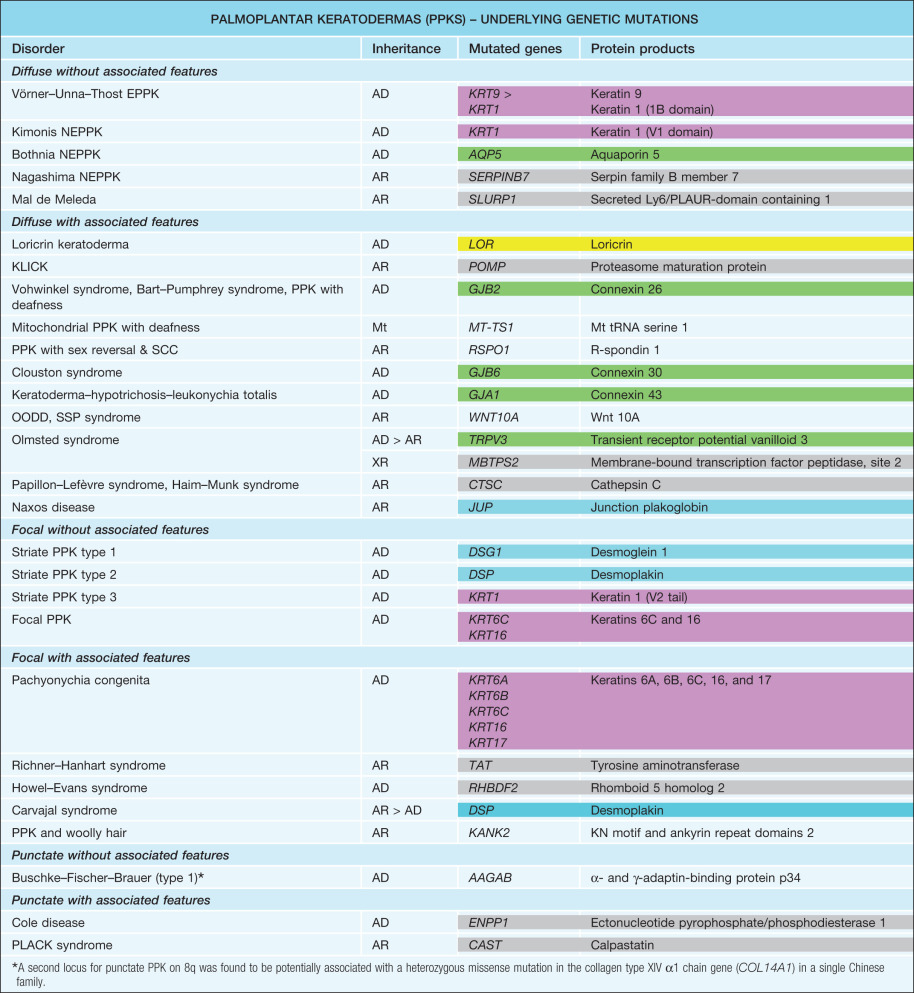
diffuse PPK – involvement of the entire palmoplantar surface, usually but not always including the central palmar skin and instep ( Fig. 58.1 )
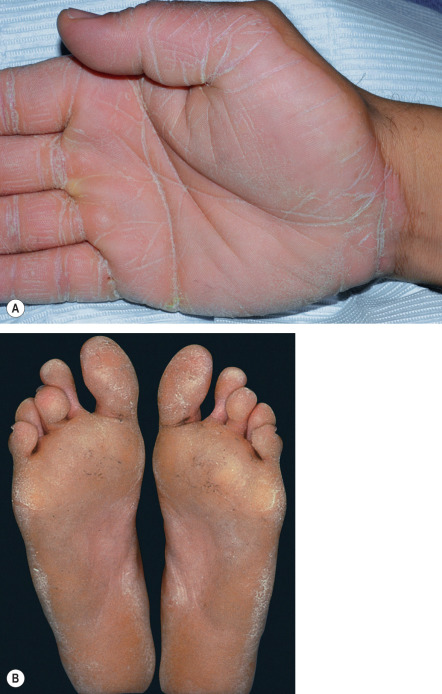
Fig. 58.1
Diffuse palmoplantar keratoderma.
Hyperkeratosis of the entire palmar (A) and plantar (B) surface with sharp demarcation.
A, Courtesy, Julie V Schaffer, MD; B, Courtesy, Alfons Krol, MD, and Dawn Siegel, MD.
- •
focal PPK – localized areas of hyperkeratosis, with two major patterns: (1) the areata/nummular type – oval lesions, located mainly over pressure points; and (2) the striate type – linear hyperkeratotic lesions, most commonly extending from the palms to the volar surface of the fingers, overlaying flexor tendons ( Fig. 58.2 ; see Fig. 58.14A )
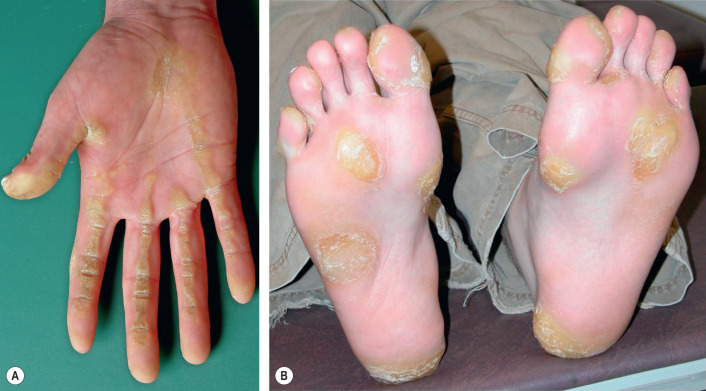
Fig. 58.2
Focal palmoplantar keratoderma.
A Striate type with linear hyperkeratosis on the palm overlaying flexor tendons. B Areata type on the soles.
B, Courtesy, Alfons Krol, MD, and Dawn Siegel, MD.
- •
punctate PPK – multiple small keratotic papules or pits (usually from removal of a keratotic plug) that are scattered or aggregated on the palms and soles ( Fig. 58.3 ).
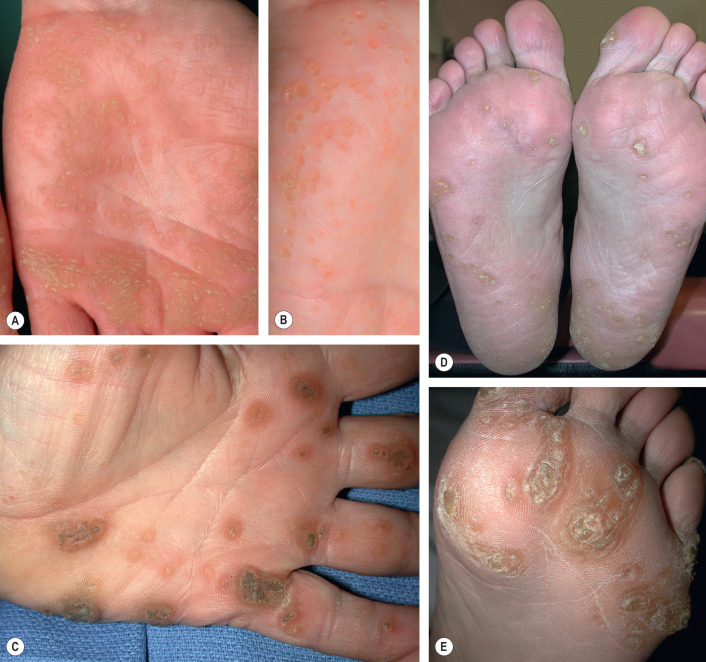
Fig. 58.3
Punctate palmoplantar keratoderma.
Keratotic papules, some coalescing to form plaques, on the palms (A–C) and soles (D, E) .
C, Courtesy, Kalman Watsky, MD.
Further clinical classification depends upon whether there are associated cutaneous and extracutaneous features (see Table 58.1 ). Many PPKs show transgrediens , which refers to extension of the hyperkeratosis onto the dorsal aspects of the fingers, toes, hands, and feet as well as the flexor aspects of the wrists and heels, either diffusely or as callosities on pressure points (e.g. the knuckles). Confluent hyperkeratosis may also extend circumferentially around entire digits. In scarring PPKs, keratotic constriction bands (pseudoainhum) can develop around digits and may lead to autoamputation ( Table 58.3 ). Additional associated findings include hyperhidrosis, maceration, blistering, malodor, pain, and fungal infections.
| GENODERMATOSES ASSOCIATED WITH DIGITAL CONSTRICTION BANDS (PSEUDOAINHUM) |
| Palmoplantar keratodermas (PPKs) |
| Other disorders of cornification |
|
| Other genodermatoses |
|
When evaluating a patient with a PPK, a thorough family history should be obtained and the entire skin surface examined as well as the mucous membranes, nails, and hair. Hearing and the ability to sweat should also be assessed. Exclusion of extracutaneous manifestations may require referral for cardiac, audiologic, ophthalmologic, or dental evaluation.
A skin biopsy for histologic examination may provide additional diagnostic clues ( Table 58.4 ). The molecular defect underlying PPKs may involve epidermal proteases as well as proteins of the keratinocyte cytoskeleton, cornified envelope, desmosome (see Fig. 56.8 ), or gap junction ( Fig. 58.4 , Table 58.5 ); the known underlying genetic mutations are summarized in Table 58.6 . Genetic analysis is sometimes helpful to establish the specific diagnosis, facilitate identification of affected family members, and enable prenatal diagnosis.
| HISTOLOGIC PATTERNS IN SELECTED HEREDITARY PALMOPLANTAR KERATODERMAS (PPKS) |
|---|
| Epidermolytic hyperkeratosis * |
|
| Orthohyperkeratosis, acanthosis, and papillomatosis |
|
| Orthohyperkeratosis with focal parakeratosis, acanthosis, and papillomatosis |
|
| Orthohyperkeratosis with parakeratosis and hypergranulosis with transitional cells |
|
| Widening of intercellular spaces and partial dehiscence of keratinocytes |
|
| Epithelia with pale cytoplasm and eosinophilic inclusions |
|
| Orthohyperkeratotic, focally parakeratotic column overlying depressed epidermis |
|
| Orthohyperkeratotic, focally parakeratotic column overlying depressed epidermis with fragmentation and loss of elastic fibers |
|
| Parakeratotic column overlaying depressed epidermis devoid of vacuolization and dyskeratosis |
|
| Parakeratotic column overlaying depressed epidermis with vacuolization and dyskeratosis |
|
* When epidermolytic hyperkeratosis is present, oral retinoid therapy can exacerbate skin fragility and should be avoided.
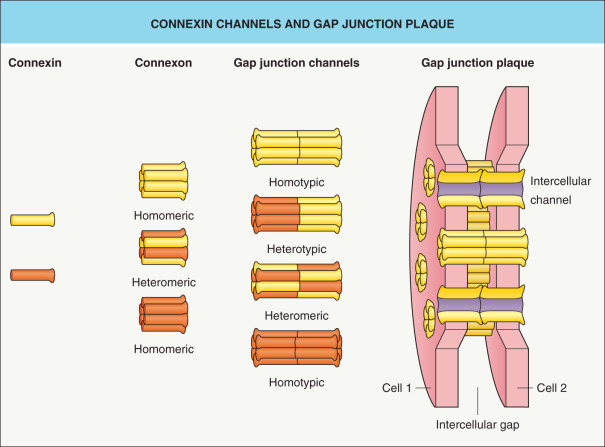
| HUMAN CONNEXIN DISORDERS | ||||
|---|---|---|---|---|
| Disorder | Inheritance | Connexin | Gene | Major organ distribution |
| Non-syndromic hearing impairment (DFNB1) | AR | Cx26 | GJB2 | Ubiquitous, including liver, cochlea, mammary gland, skin and appendages, mucous membranes, pancreas, kidney, intestine, lung |
| Non-syndromic hearing impairment (DFNA3) | AD | Cx26 | GJB2 | |
| Palmoplantar keratoderma and hearing impairment, including Vohwinkel syndrome | AD | Cx26 | GJB2 | |
| Bart–Pumphrey syndrome | AD | Cx26 | GJB2 | |
| Keratitis–ichthyosis–deafness (KID) syndrome & hystrix-like–ichthyosis–deafness (HID) syndrome | AD | Cx26 (Cx30) * | GJB2 (GJB6 ) * | |
| Porokeratotic adnexal ostial nevus (PAON) | Mosaic | Cx26 | GJB2 | |
| Non-syndromic hearing impairment (DFNA3) | AD | Cx30 | GJB6 | Brain, cochlea, skin |
| Clouston syndrome | AD | Cx30 | GJB6 | |
| Erythrokeratodermia variabilis | AD≫AR | Cx30.3 | GJB4 | Skin, kidney |
| Erythrokeratodermia variabilis | AD≫AR | Cx31 | GJB3 | Skin, eye, placenta, kidney, testes, cochlea, Schwann cells |
| Non-syndromic hearing impairment | AR≫AD | Cx31 | GJB3 | |
| Peripheral sensory neuropathy and hearing impairment | AD | Cx31 | GJB3 | |
| Charcot–Marie–Tooth disease | X-linked dominant | Cx32 | GJB1 | Brain, Schwann cells, liver, thyroid, kidney, pancreas, uterus |
| Polymorphism may be a marker for atherosclerotic plaque development | Cx37 | GJA4 | Gonads, lung, endothelium, heart, skin | |
| Oculo-dento-digital dysplasia | AD | Cx43 | GJA1 | Ubiquitous, including eyes, teeth, musculoskeletal, brain, heart, skin |
| Erythrokeratodermia variabilis et progressiva | AD | Cx43 | GJA1 | |
| Keratoderma-hypotrichosis-leukonychia totalis | AD | Cx43 | GJA1 | |
| ILVEN | Mosaic | Cx43 * | GJA1 * | |
| Syndactyly, type III | AD | Cx43 | GJA1 | |
| Hypoplastic left heart syndrome | Oligogenic | Cx43 | GJA1 | |
| Cataract, zonular pulverulent-3 | AD | Cx46 | GJA3 | Lens, heart, kidney, peripheral nervous system |
| Atrial fibrillation, familial 11 | AD | Cx40 | GJA5 | Heart |
| Hereditary lymphedema | AD | Cx47 | GJC2 | Lymphatic vessels, CNS |
| Hypomyelinating leukodystrophy | AR | Cx47 | GJC2 | |
| Cataract, zonular pulverulent-1 | AD | Cx50 | GJA8 | Lens, cornea, heart |
Hereditary Keratodermas
Diffuse Palmoplantar Keratoderma Without Associated Features (Isolated, Non-Syndromic)
Diffuse Epidermolytic Palmoplantar Keratoderma (EPPK)
▪ Unna–Thost disease ▪ Vörner disease ▪ Vörner–Unna–Thost type PPK ▪ Keratosis palmoplantaris diffusa Vörner–Unna–Thost
History
In 1880, Thost described a family with diffuse non-transgrediens PPK. This was followed by Unna’s description of a clinically identical, autosomal dominant PPK in two families. Although “Unna–Thost type” has been used to designate non-epidermolytic PPK (NEPPK), in 1992, Küster et al. reviewed the family described by Thost and found the histologic features of epidermolytic hyperkeratosis, which characterize the “Vörner type” of PPK. Subsequently, after keratin mutations were reported in epidermolytic PPK (EPPK), identical mutations were found in the descendants of the family studied by Thost . This confusion underscores the need for careful histologic examination and sometimes multiple biopsy specimens to identify epidermolytic hyperkeratosis.
Epidemiology
EPPK is likely the most common form of diffuse keratoderma, with an estimated incidence of ≥4.4 per 100 000 in Northern Ireland .
Pathogenesis
Mutations in KRT1 and KRT9 underlie diffuse EPPK. The expression of keratin 9 is limited to the suprabasal cell layers of palmar and plantar skin. In patients with diffuse EPPK, the majority of keratin 9 mutations are localized to a very small area within this gene that encodes the helix initiation motif in the 1A domain (see Ch. 56 ). Such mutations are highly disruptive to keratin filament assembly, causing tonofilament clumping and cytolysis that result in blistering as well as hyperkeratosis.
Within palmoplantar skin, keratin 9 is thought to partner with keratin 1. Of note, mutations in keratin 1 and 10 are associated with epidermolytic ichthyosis (EI; formerly bullous congenital ichthyosiform erythroderma), in which epidermolytic hyperkeratosis is seen histologically (see Ch. 57 ). PPK in EI is predictive of a mutation in keratin 1, which is the only type II keratin expressed in palmoplantar skin. In contrast, EI due to keratin 10 mutations rarely has involvement of palmoplantar skin, likely reflecting compensation by keratin 9, which is also a type I keratin. The majority of mutations in EI patients are in the critical 1A and 2B domains of the keratin molecule . However, when keratin 1 defects underlie diffuse EPPK alone, including the “tonotubular“ subtype, the mutations are typically in the beginning of the 1B domain . Although keratin 1 mutations have been reported in some families with the Greither type of EPPK, other Greither variants with NEPPK not associated with keratin 1 mutations have also been described. Of note, keratin 1 mutations have also been identified in patients with focal PPK (see below).
Clinical features
The palmoplantar skin is initially red, followed by the appearance of thick, yellow hyperkeratosis by 3–4 years of age. In adults, there is confluent hyperkeratosis with a smooth, waxy surface and sharply demarcated erythematous border, sparing the dorsal aspects of the palms and soles ( Fig. 58.5 ). Blistering and fissuring are not common but may occur during childhood or during oral retinoid therapy. Knuckle pads and thickened nails are occasionally present . Disabling pain, especially of the palms, can occur in the “tonotubular” EPPK subtype . The Greither type (PPK transgrediens and progrediens) presents with limited transgredient lesions, e.g. overlying the Achilles tendon, elbows, and knees.
Pathology
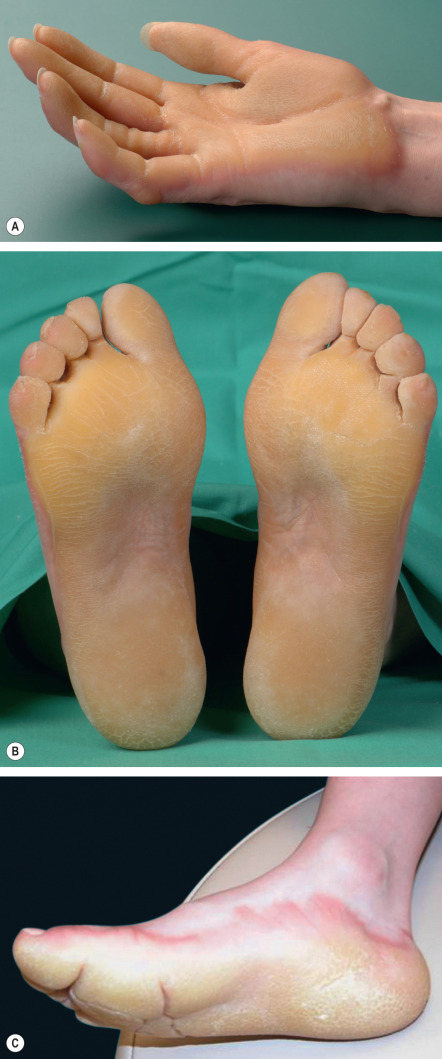
EPPK shows the typical pattern of epidermolytic hyperkeratosis, which is characterized by vacuolated suprabasal keratinocytes with round or ovoid eosinophilic inclusions that represent large tonofilament aggregates at an ultrastructural level . In the “tonotubular” subtype, electron microscopy shows whorls of keratin containing tubular structures . Histologic findings also include coarse keratohyalin granules, acanthosis, and marked orthohyperkeratosis. Intraepidermal blistering and a mild superficial dermal inflammatory infiltrate may result from keratinocyte cytolysis ( Fig. 58.6A ). The findings of epidermolytic hyperkeratosis are often subtle and patchy, requiring a careful search and often multiple biopsy specimens to enable their identification.
Differential diagnosis
EI can be distinguished clinically by the cutaneous involvement beyond the palms and soles. EPPK and NEPPK can be differentiated histologically by the absence of epidermolytic hyperkeratosis in the latter; they are not distinguishable clinically, although the erythematous border of EPPK tends to be more pronounced.
Management
Factors to consider when selecting treatment for diffuse PPK include the severity of symptoms, degree of hyperkeratosis, and age of the patient. Mechanical debridement with a blade or dental drill is useful for troublesome areas, followed by application of a keratolytic agent to help avoid fissure formation. Options for keratolytic therapy include 50% propylene glycol in water under plastic occlusion several nights per week, lactic acid- and urea-containing creams and lotions, and salicylic acid 4–6% in petrolatum; the latter should not be applied to large areas in young children to avoid salicylism from systemic absorption (see Ch. 129 ). Oral retinoids may improve the hyperkeratosis, but even low doses can lead to excessive peeling and increased fragility of volar skin in EPPK . Topical calcipotriene (calcipotriol) has reportedly been helpful .
Diffuse Non-epidermolytic Palmoplantar Keratoderma (NEPPK)
Diffuse NEPPK represents a heterogeneous group of non-syndromic forms of PPK that do not show epidermolytic hyperkeratosis histologically. This category includes Bothnia , Kimonis , and Nagashima types of PPK as well as Mal de Meleda . The Greither type (PPK transgrediens and progrediens) lacks a clear definition and is heterogeneous, with both epidermolytic and non-epidermolytic variants (see EPPK above) . The Gamborg Nielsen (Norrbotten) type is currently regarded as a variant of Mal de Meleda .
Epidemiology and pathogenesis
An autosomal dominant form of NEPPK described in a family from Bothnia in Northern Sweden and three English pedigrees is due to heterozygous missense mutations in AQP5 , which encodes aquaporin 5. Aquaporins are transmembrane proteins that allow the osmotic movement of water across the cell membrane. Aquaporin 5 is expressed by keratinocytes in the granular layer, and the gain-of-function mutations that cause Bothnia-type NEPPK lead to increased keratinocyte water uptake rather than transepidermal water loss .
The form of NEPPK described by Kimonis is caused by a heterozygous mutation in a highly conserved lysine residue in the V1 domain of keratin 1 that does not result in epidermolytic hyperkeratosis .
Nagashima -type NEPPK, an autosomal recessive disorder almost exclusively observed in Asia, is caused by biallelic nonsense mutations in SERPINB7. This gene encodes the ser ine p rotease in hibitor family B member 7, which has an unknown protease target in the skin .
Mal de Meleda is an autosomal recessive condition that was first described in inhabitants of the island of Mljet (Meleda) off the Dalmatian coast. It is caused by biallelic mutations in SLURP1 ( s ecreted l eukocyte antigen-6/ u rokinase-type plasminogen activator related p rotein 1 ), which is expressed in the granular layer of the epidermis . SLURP1 acts as a ligand of the α7-nicotinic acetylcholine receptor and plays a role in epidermal differentiation. Heterozygous female carriers may have a mild clinical phenotype.
Clinical features
The clinical findings and time course of the different types of NEPPK are overall similar to those of EPPK (see above). The hyperkeratosis is variably thick and hyperhidrosis is common, with frequent dermatophyte infections and pitted keratolysis. The Bothnia type features a characteristic white spongy appearance upon exposure to water .
In Mal de Meleda, progressive transgredient hyperkeratosis begins during early infancy and becomes severe, impairing the mobility of hands and resulting in hyperhidrotic maceration, severe malodor, and fissures ( Fig. 58.7 ). Fungal superinfection is common, and hyperkeratosis of the fingers may lead to sclerodactyly and digital constrictions (pseudoainhum). The elbows, knees, wrists, and ankles as well as the dorsal surfaces of the hands and feet can be involved, and the nails show thickening, koilonychia, and subungual hyperkeratosis. Angular cheilitis and involvement of the lips and perioral skin may be observed . Hyperpigmented macules, melanoma, and Bowen disease within the keratotic areas have been reported .
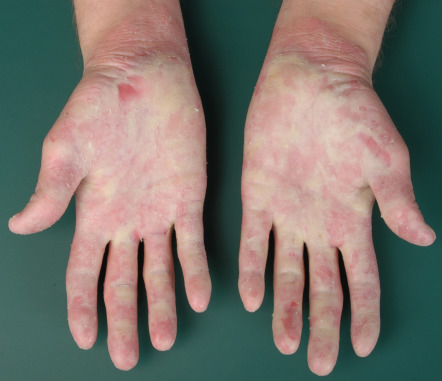
Differential diagnosis
In addition to diffuse EPPK, exfoliative ichthyosis and aquagenic palmoplantar keratoderma may be diagnostic considerations.
Pathology
Histologically, palmoplantar skin shows orthohyperkeratosis, hypergranulosis or normogranulosis, and acanthosis ( Fig. 58.6B ). A moderate perivascular lymphocytic infiltrate may be present. In Mal de Meleda, papillomatosis is a characteristic finding. PAS staining should be performed to exclude infection by dermatophytes.
Management
Stronger keratolytic therapy may be helpful, such as 5–10% salicylic acid in white soft paraffin, or 5–6% salicylic acid in 70% propylene glycol gel; occlusion for a few nights per week enhances efficacy, and mechanical debridement can also be performed. Low-dose acitretin (0.2–0.5 mg/kg daily) may be considered, especially for patients with functional impairment. Fungal superinfection and bacterial overgrowth should be treated. Responses to oral erythromycin and topical tacrolimus have been described in the Bothnia and Nagashima types, respectively . Excision of hyperkeratotic skin with split-thickness skin grafting represents an option to relieve functional impairment .
Diffuse Palmoplantar Keratodermas With Ichthyosis
Loricrin keratoderma and k eratosis l inearis– i chthyosis c ongenita–sclerosing k eratoderma (KLICK) have distinctive skin findings beyond the palms and soles. PPK is also a component of other ichthyoses that are discussed in Chapter 57 (see Table 58.2 ).
Loricrin Keratoderma
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


