Abstract
The clinicopathologic features of the following entities are reviewed: systemic-onset juvenile idiopathic arthritis (sJIA), adult-onset Still disease, relapsing polychondritis, Sjögren syndrome, mixed connective tissue disease (MCTD), and rheumatoid arthritis, as well as autoinflammatory diseases. Although all these disorders represent systemic multi-organ diseases, the pathomechanisms differ. Disorders such as sJIA, adult-onset Still disease, and autoinflammatory diseases are related to abnormalities in the innate immune system, i.e. neither autoantibodies nor autoreactive T cells play an important role. Other disorders, including rheumatoid arthritis, Sjögren syndrome and MCTD, are characterized by autoimmune-mediated tissue damage and the presence of characteristic autoantibodies such as anti-Ro and/or anti-La in Sjögren syndrome, anti-ribonucleoprotein in MCTD, or anti-cyclic citrullinated peptide in rheumatoid arthritis.
Keywords
systemic-onset juvenile idiopathic arthritis (Still disease), adult-onset Still disease, relapsing polychondritis, Sjögren syndrome, mixed connective tissue disease, rheumatoid arthritis, autoimmune connective tissue disease, autoinflammatory diseases, interleukin-1 inhibitors.
Although all the disorders in this chapter represent inflammatory systemic diseases, there are two major groups based upon underlying pathomechanisms. Disorders such as systemic-onset juvenile idiopathic arthritis (Still disease) and inherited autoinflammatory diseases are related to abnormalities in the innate immune system whereas other disorders, including rheumatoid arthritis and Sjögren syndrome, are characterized by autoimmune-mediated tissue damage via autoreactive antigen-specific T cells and the presence of characteristic autoantibodies.
Systemic-Onset Juvenile Idiopathic Arthritis (Still Disease)
▪ Juvenile idiopathic arthritis: juvenile rheumatoid arthritis (JRA); juvenile chronic arthritis
- ▪
There are several types of juvenile idiopathic arthritis, including systemic-onset, polyarticular (either rheumatoid factor [RF]-positive or RF-negative) and oligoarticular
- ▪
Systemic-onset juvenile idiopathic arthritis, also known as Still disease, is characterized by high spiking fevers, lymphadenopathy, hepatosplenomegaly, transient cutaneous eruptions of erythematous macules and papules (often accompanying the fevers and associated with Koebner phenomena), and arthritis
Introduction
Juvenile idiopathic arthritis (JIA) is classified into seven major types ( Table 45.1 ) , including systemic-onset, rheumatoid factor (RF)-positive polyarticular, RF-negative polyarticular, and oligoarticular. Systemic-onset JIA (sJIA), also known as Still disease, is characterized by an evanescent, erythematous eruption that typically accompanies spiking fevers. Cutaneous manifestations are less common in the other types, although the RF-positive subgroup of polyarticular JIA can present with rheumatoid nodules and other skin findings similar to those seen in adults with rheumatoid arthritis (see below), and nearly all patients with psoriatic arthritis have cutaneous and/or nail psoriasis (see Ch. 8 ).
| CLASSIFICATION OF JUVENILE IDIOPATHIC ARTHRITIS | |
|---|---|
| Systemic onset (15–20%) (Still disease) | Features required for diagnosis
Other features
|
| RF-negative polyarticular arthritis (15%) |
|
| RF-positive polyarticular arthritis (5%) |
|
Oligo/pauciarticular arthritis (50%):
|
|
| Enthesitis-related arthritis (5–10%) |
|
| Psoriatic arthritis (~5%) |
|
| Other arthritis |
|
* Onset may be delayed for months to years.
† The persistent oligoarticular arthritis subtype never affects more than 4 joints, whereas the extended oligoarticular arthritis subtype affects a cumulative total of ≥5 joints after the first 6 months of disease.
History
In 1897, George Still, an English pediatrician, first described a chronic arthritis in children that was associated with fever, lymphadenopathy, and organomegaly.
Epidemiology
JIA is the most common rheumatic disease in childhood, and its overall prevalence is estimated to be 0.2–1 per 1000 children (by definition ≤16 years of age) . sJIA comprises 15–20% of all cases of JIA and can occur at any time from infancy through adolescence, with a mean age at onset of 6 years. Whereas most other types of JIA have a female predominance, sJIA affects both sexes equally. A monogenic form of sJIA with an underlying mutation in LACC1 has been described; its protein product regulates metabolism within macrophages .
Pathogenesis
Recent insight into the pathogenesis of sJIA suggests that it is better classified as an autoinflammatory rather than an autoimmune disease. The inflammation characteristic of this disease appears to result from activation of innate immune responses rather than adaptive phenomena characterized by antigen-specific, lymphocyte-driven immune responses (see Ch. 4 ). Dysregulated production of interleukin (IL)-1 appears to play a critical role in the pathogenesis of sJIA. When peripheral blood mononuclear cells (PBMCs) from healthy individuals are exposed to sera from patients with sJIA, there is upregulation of gene expression for the innate inflammatory cytokines IL-1α and IL-1β (as well as upregulation of other innate immunity genes) . Of note, the administration of IL-1 antagonists (e.g. anakinra, rilonacept, canakinumab) can dramatically reduce disease activity in a subset of patients with sJIA (see below) .
The dysregulated IL-1 production may be due to inflammasome activation (see Fig. 4.2 ) and/or dysregulation of the alternative secretory pathway downstream of caspase-1 that is responsible for the production of the calcium-binding proteins S100A8, S100A9 and S100A12; the latter are secreted during activation of neutrophils and monocytes . Patients with sJIA have exceptionally high serum levels of S100A8, S100A9 and S100A12, and these proteins do exhibit proinflammatory effects on endothelial cells and leukocytes.
At the onset and during flares of sJIA, vascular endothelial cells express leukocyte adhesion molecules, leading to a perivascular infiltrate of neutrophils and activated monocytes that secrete proinflammatory cytokines including tumor necrosis factor-α (TNF-α), IL-1 and IL-6 . Many of the clinical features of sJIA can be explained by the effects of these cytokines. For example, IL-1 stimulates granulopoiesis in the bone marrow and activates thermoregulatory functions of the hypothalamus to cause fever. The chronic overexpression of IL-6 observed in sJIA increases osteoclastogenesis and reduces osteoblast activity. IL-6 also stimulates hepatocytes and induces the production of acute phase reactants such as C-reactive protein .
Clinical Features
Daily high fevers (usually above 38.9°C) that typically occur in the late afternoon or early evening are characteristic of sJIA. The arthritis is frequently polyarticular, and the joints most commonly affected are the knees, ankles and hips, followed by the small joints of the hands. A subgroup of patients may develop destructive joint disease similar to that seen in RF-positive polyarticular arthritis (see Table 45.1 ).
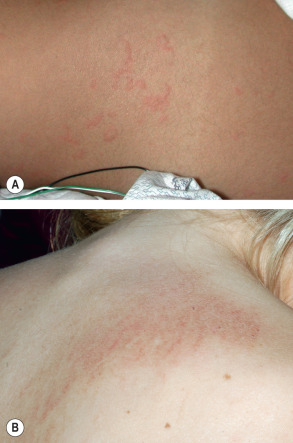
An exanthem is present in up to 90% of patients with the acute febrile presentation of sJIA, and it is often preceded by or accompanied by arthralgias. Although arthritis usually develops during the first few months of the disease course, in approximately one-quarter of patients the exanthem begins prior to the onset of arthritis, occasionally predating joint involvement by several years. The eruption is typically transient, non-pruritic and erythematous, and it coincides with fevers ( Fig. 45.1A ). There is a predilection for the axillae and waist, and linear lesions secondary to the Koebner phenomenon are often observed. Other less common cutaneous features include persistent plaques, which also may be linear ( Fig. 45.1B ), and periorbital edema and erythema . Rheumatoid nodule-like lesions that favor extensor surfaces have been described in the setting of methotrexate therapy for sJIA . Additional systemic features are outlined in Table 45.1 . Although there are no laboratory findings specific for sJIA, leukocytosis, granulocytosis, thrombocytosis, elevated hepatic enzymes, an elevated ESR, and a polyclonal gammopathy are commonly observed; ANA and RF are rarely present. Serum ferritin levels are elevated and typically become even higher in the setting of macrophage activation syndrome (see below).
Up to 10% of children may develop a serious and potentially lethal complication, the macrophage activation syndrome (MAS), characterized by fever, cytopenias, liver dysfunction, coagulopathy, hypofibrinogenemia, hypertriglyceridemia, and very high serum levels of ferritin (see Table 91.1 ). Increased serum IL-18 levels and depressed NK cell cytolytic function are also observed in patients with sJIA who develop MAS.
Pathology
The evanescent exanthem is characterized by a perivascular and interstitial neutrophil-dominant mixed infiltrate, a reaction pattern also seen in autoinflammatory diseases (see below) and referred to as a “neutrophilic urticarial dermatosis” . A variable number of dyskeratotic keratinocytes, located primarily in the upper epidermal layers, is also a typical finding.
Differential Diagnosis
The differential diagnosis of a transient exanthem associated with fever and arthritis includes rheumatic fever, urticarial vasculitis, serum sickness-like reaction, and hereditary periodic fever syndromes ( Table 45.2 ). The characteristic cutaneous findings in rheumatic fever are erythema marginatum (see Ch. 19 ) and urticarial lesions; rheumatic fever nodules are unusual. Patients with urticarial vasculitis may have arthralgias and fever; however, the urticarial lesions persist for greater than 24 hours, and they often resolve with purpura or postinflammatory hyperpigmentation. Histologic examination of recent-onset lesions demonstrates leukocytoclastic vasculitis.
| HEREDITARY PERIODIC FEVER SYNDROMES | ||||||
|---|---|---|---|---|---|---|
| FMF | HIDS | TRAPS * | Cryopyrin-associated periodic syndromes (CAPS) | |||
| MWS | FCAS † (1 and 2) | NOMID (CINCA) | ||||
| Ethnicity | Sephardic Jewish, Arab, Turkish, Italian, Armenian | Predominantly Dutch, northern European | Any ethnic group | Any ethnic group | Any ethnic group | Any ethnic group |
| Inheritance | AR | AR | AD | AD | AD | AD |
| Gene | MEFV | MVK ‡ | TNFRSF1A | NLRP3 (CIAS1) | NLRP3 (CIAS1); NLRP12 | NLRP3 (CIAS1) |
| Chromosome | 16p13.3 | 12q24 | 12p13 | 1q44 | 1q44 | 1q44 |
| Protein | Pyrin | Mevalonate kinase | TNF receptor-1A | Cryopyrin | Cryopyrin; monarch-1 | Cryopyrin |
| Attack length | 1–3 days | 3–7 days | Often >7 days | 1–2 days | Minutes–3 days | Continuous + flares |
| Mucocutaneous lesions | Erysipeloid erythema and edema | Erythematous macules and edematous papules, which may become purpuric; occasional oral and vaginal ulcers | Erythematous patches and edematous plaques (often annular or serpiginous); later ecchymotic in appearance; rarely oral ulcers | Urticarial papules and plaques | Cold-induced urticarial papules and plaques | Urticarial papules and plaques; occasional oral ulcers |
| Distribution of skin lesions | Favor lower leg, foot | Widespread on face, trunk and extremities | Migrate distally on an extremity with underlying myalgia; may be more widespread | Widespread on face, trunk and extremities | Extremities > trunk, face | Widespread on face, trunk and extremities |
| Abdominal pain and serositis | Peritonitis > pleuritis > pericarditis | Abdominal pain, but rarely serositis | Peritonitis > pleuritis, pericarditis | Abdominal pain, but rarely serositis | Rare | Rare |
| Musculoskeletal findings | Monoarthritis > exercise-induced myalgia | Arthralgia > oligoarticular arthritis > myalgia | Migratory myalgia > arthralgia > monoarthritis | Myalgia (“lancinating limb pain”), arthralgia > large-joint oligoarticular arthritis | Arthralgia > myalgia | Epiphyseal and patellar overgrowth, arthritis, deforming arthropathy |
| Ocular findings | Uncommon | Uncommon | Periorbital edema, conjunctivitis, rarely uveitis | Conjunctivitis, episcleritis, optic disc edema | Conjunctivitis | Conjunctivitis, uveitis, optic disc edema, blindness |
| Neurologic findings | Rarely aseptic meningitis | Headache | Headache | Sensorineural hearing loss; headache | Headache | Sensorineural hearing loss; aseptic meningitis, seizures |
| Other clinical findings | Acute scrotal swelling; splenomegaly | Cervical LAN, HSM | Scrotal pain; splenomegaly, occasional LAN | LAN, HSM; dysmorphic facies – frontal bossing, protruding eyes | ||
| Amyloidosis | Most common in M694V homozygotes | Rare | ~15% of cases | ~25% of cases | Uncommon | Late complication |
| Dermal infiltrate in typical skin lesions | Neutrophils | Neutrophils and/or lymphocytes; mild vasculitis common | Lymphocytes, monocytes and a few neutrophils | Neutrophils and/or lymphocytes (sparse) | Neutrophils (perivascular) | Neutrophils (perivascular + periadnexal) |
| Cutaneous vasculitis | LCV/HSP (5–10%), PAN (~1%) | LCV/HSP | Lymphocytic small vessel (rare) | |||
| Laboratory abnormalities § | Low C5a inhibitor in serosal fluids | High serum IgD ‖ (>100 IU/ml) and IgA1; mevalonate in urine during attacks; low lymphocyte mevalonate kinase | Low serum soluble TNF receptor-1 (<1 ng/ml between attacks) | |||
| Treatment | Colchicine prophylaxis; NSAIDs, TNF inhibitors, thalidomide, herbal remedies | Corticosteroids for acute attacks; anakinra; TNF inhibitors (e.g. etanercept); simvastatin | Corticosteroids; TNF inhibitors (e.g. etanercept) | Corticosteroids; IL-1/IL-1R antagonists: rilonacept, canakinumab, anakinra | IL-1/IL-1R antagonists | IL-1/IL-1R antagonists |
* Includes familial Hibernian fever.
† Also referred to as familial cold urticaria.
‡ Allelic with mevalonic aciduria, which is characterized by dysmorphology, psychomotor retardation and progressive cerebellar ataxia as well as periodic fevers and other features of HIDS.
§ Genetic analysis can be performed to confirm the diagnosis.
‖ IgD levels are occasionally normal; elevated IgD may also be observed in FMF and TRAPS.
Additional disorders to consider include other autoimmune connective tissue diseases; leukemia (which may present with fevers and musculoskeletal complaints prior to the development of diagnostic abnormalities in the peripheral blood) as well as other malignancies (e.g. lymphoma); and infections such as parvovirus B19, malaria, and rat bite fever.
Treatment
The course and prognosis of sJIA is variable. In ~40–50% of patients, the arthritis resolves completely. However, approximately one-half of the children may have a chronic course that includes persistent arthritis and systemic complications such as macrophage activation syndrome (see above), hepatitis, pericarditis, and rarely amyloidosis (see Table 45.1 ). Patients with symptoms that persist for longer than 6 months carry a worse prognosis.
Mild articular or extra-articular disease can be treated with nonsteroidal anti-inflammatory drugs (NSAIDs). Moderate or severe disease often requires the use of systemic corticosteroids with or without adjuvant drugs such as methotrexate. Inhibitors of TNF-α have been increasingly used as adjuvant or monotherapy; however, these agents are less effective in patients with sJIA than in those with other types of JIA . Potent corticosteroid-sparing or immunomodulating agents, such as abatacept, azathioprine and leflunomide, are required infrequently . Thalidomide is also a therapeutic option for children with sJIA. It suppresses the activity of cytokines (e.g. TNF-α and IL-6) that have been shown to play a role in the fever and malaise associated with sJIA. In clinical trials, antagonists of the IL-1 receptor (e.g. anakinra, rilonacept, canakinumab) and antagonists of the IL-6 receptor (e.g. tocilizumab) have demonstrated efficacy in sJIA ; as a result, they may become first-line therapies. Hematopoietic stem cell transplantation has been successfully performed in children who have failed to respond to combinations of medications .
Adult-Onset Still Disease
▪ Still disease in the adult ▪ Adult Still disease
- ▪
A characteristic feature is recurrent episodes of high spiking fevers that often occur in the late afternoon
- ▪
An asymptomatic macular exanthem accompanies the fevers, is salmon-pink in color, and frequently demonstrates the Koebner phenomenon
- ▪
Arthritis with subsequent carpal ankylosis is a distinctive feature
Introduction
Adult-onset Still disease can be a severe debilitating systemic illness that is often difficult to diagnose. The vast majority of these patients have an exanthem, and this can be the most distinctive sign of the disorder.
Epidemiology
Adult-onset Still disease primarily affects young adults, with an onset before the age of 30 years in the majority of patients. Rarely it occurs in individuals over the age of 60 years. Women are affected slightly more often than men.
Pathogenesis
Although the etiology of adult-onset Still disease is unknown, the underlying pathogenesis likely overlaps with that of sJIA (see above). The disease has been linked with a number of HLA antigens, including HLA-B14, -B17, -B18, -B35, -Bw35, -Cw4, -DR2, -DR7, -DR4, and -Dw6, suggesting a genetic component . It has been hypothesized to occur in a susceptible individual as a reactive condition triggered by a variety of infectious agents, including viruses (rubella, mumps, echovirus 7, cytomegalovirus, EBV, parainfluenza, influenza A, coxsackievirus B4, adenovirus, human herpesvirus 6, parvovirus B19, hepatitis B, hepatitis C) and bacteria ( Mycoplasma pneumoniae , Chlamydophila [ Chlamydia ] pneumoniae , Yersinia enterocolitica 3 and 9, Brucella abortus , Borrelia burgdorferi ) . To date, however, no infectious agent has been implicated consistently.
The role of Th1 cytokines in the pathogenesis of adult-onset Still disease has been the subject of more recent investigation. Of note, serum levels of IL-2, IL-6, IL-18, TNF-α and IFN-γ are elevated in patients with the disease .
Clinical Features
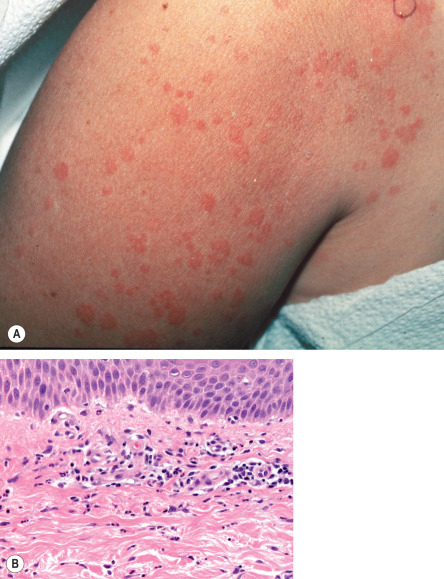
The onset of the disease is frequently heralded by a sore throat and constitutional symptoms including arthralgias and myalgias. Fever is a prominent feature; it is usually above 39°C and has a spiking pattern, often occurring in the late afternoon or early evening and resolving within several hours. The skin eruption is asymptomatic and transient, occurring in association with the fever spikes. It is most often macular with a characteristic salmon-pink color ( Fig. 45.2A ), favors sites of pressure, and can exhibit the Koebner phenomenon. Although the trunk is most commonly involved, lesions can appear on the extremities, including the palms and soles. Violaceous to reddish brown, scaly, persistent papules and plaques can also occur, and were observed in more than half of patients with adult-onset Still disease in a Taiwanese series . Persistent erythematous eyelid edema and flagellate erythema are rare manifestations .
Depending upon the series, arthritis occurs in 65–100% of patients, and most often it affects the knees, wrists and ankles, although other joints may also be involved. The pattern is usually symmetric, and most patients ultimately develop joint pain associated with the fever spikes. A distinctive feature of the arthritis of adult-onset Still disease is carpal ankylosis, which results in minimal pain but a limited range of motion in the carpal joints. Similar ankylosis may occur in the proximal and distal interphalangeal joints as well as the cervical spine. The metacarpophalangeal (MCP) joints are usually spared.
There may be hepatomegaly, but splenomegaly is less common. Involvement of the lungs (pleuritis, pleural effusions, pulmonary fibrosis, pulmonary arterial hypertension), heart (pericarditis, myocarditis, cardiac tamponade), and kidneys (nephritis) is rare.
Laboratory findings in adult-onset Still disease are similar to those of sJIA and include elevations in the C-reactive protein level, ESR, and platelet count. Leukocytosis and anemia are common, as are abnormalities in liver function tests. There are often significant elevations in serum ferritin levels, sometimes >4000 mg/ml, and ferritin levels may correlate with disease activity ; a low circulating level of glycosylated ferritin (fraction <20%) is characteristic. ANA and RF are either negative or, if present, in a low titer.
Pathology
Histopathologic changes are indistinguishable from those of the juvenile form of the disease (see above) and include an interstitial and perivascular neutrophil-dominant mixed infiltrate ( Fig. 45.2B ) .
Differential Diagnosis
Adult-onset Still disease should always be considered in an adult with fever of unknown origin (defined as fever >101°F [38.3°C] on several occasions over a period of at least 3 weeks or uncertain diagnosis after 1 week of hospitalization). The differential diagnosis is similar to that of sJIA (see above). Additionally, Schnitzler syndrome should be considered in an adult patient with recurrent fevers, arthralgia, and an urticarial eruption (see Ch. 18 ). In addition to non-pruritic urticaria, patients have recurrent fevers, bone pain (lower extremity, iliac and vertebral due to hyperostosis), and a monoclonal IgM gammopathy. Angioedema is observed in about 15% of patients with Schnitzler syndrome, and lymphoplasmacytic malignancies in 10–15% . Histologic features are similar to those of adult-onset Still disease, i.e. a neutrophilic urticarial dermatosis , but the neutrophilic infiltrate may be more dense.
Treatment
Although some patients respond to high-dose aspirin or NSAIDs, the majority require oral corticosteroids (e.g. 40–60 mg prednisone daily) to control acute systemic features. When corticosteroids cannot be tapered, methotrexate is the most commonly employed second-line therapy. As in sJIA, biologic agents that inhibit the IL-1 receptor or IL-6 receptor (e.g. tocilizumab) appear to be promising . A therapeutic response to TNF-α inhibitors has also been reported, especially in those patients with severe chronic polyarticular disease. In addition, there have been reports of rituximab leading to improvement of refractory disease .
Relapsing Polychondritis
▪ Atrophic polychondritis ▪ Systemic chondromalacia
- ▪
A prominent clinical feature is erythema, swelling and pain of the cartilaginous portion of the ear, followed by destruction of the cartilage
- ▪
Additional findings include nasal chondritis, which may result in a saddle nose deformity, as well as arthritis of the joints of the central chest
- ▪
The most serious complications result from ocular, renal, and respiratory tract involvement; some of the patients develop a myelodysplastic syndrome
Introduction
Relapsing polychondritis is an uncommon inflammatory disorder with a suspected autoimmune origin that primarily affects cartilaginous structures. The diagnosis is usually established based upon the presence of: (1) histologically confirmed chondritis in two of the following three sites: auricular, nasal, or laryngotracheal cartilage; or (2) chondritis in one of the aforementioned sites plus at least two other features, including: ocular inflammation, vestibular dysfunction, hearing loss, and seronegative inflammatory arthritis. Relapsing polychondritis has been associated with other autoimmune disorders (25–30% of patients) and with myelodysplastic syndromes. Dermatologic manifestations may be the initial presenting sign and early diagnosis and treatment can prevent later complications, e.g. ascending aortitis, glomerulonephritis.
History
The disease was first described in 1923 by Jaksch-Wartenhorst and was called “polychondropathia”. It was later renamed “relapsing polychondritis” due to its episodic nature.
Epidemiology
Relapsing polychondritis is most common in Caucasians, but has been reported in other races. In 80% of cases, the onset is between the ages of 20 and 60 years. Men and women are equally affected.
Pathogenesis
Although the etiology of relapsing polychondritis is unknown, the pathogenesis seems to involve an immunologic reaction against type II collagen . However, circulating antibodies against type II collagen are present in fewer than half of affected individuals, and antibodies against types IX and XI collagen have also been described . The clinical usefulness of these antibodies in the diagnosis, prognosis or monitoring of disease activity or the response to therapy is unclear. That said, transmission of relapsing polychondritis to a neonate from an affected mother has been reported (with subsequent recovery of the infant), lending support to an antibody-mediated pathogenesis. Antibodies to matrilin-1, an extracellular matrix protein located in auricular, septal, tracheal and sternal cartilage, may also play a role in the immune response seen in this disease . Additionally, there is a positive association with HLA-DR4, while the extent of organ involvement has been negatively associated with HLA-DR6.
Clinical Features
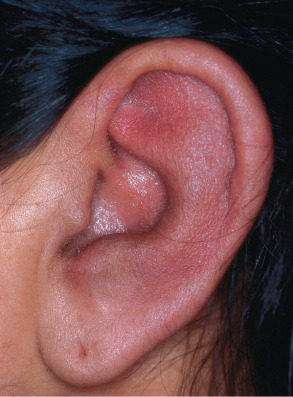
The most prominent clinical feature is erythema, swelling and pain of the cartilaginous portion of the auricle , sparing the earlobe ( Fig. 45.3 ). Symptoms can persist for several days to weeks and may encroach on the external auditory meatus, compromising hearing. Chronic inflammation leads to destruction of the cartilage, leaving the ear unsupported and scarred. During the course of their disease, 90% of patients will develop auricular involvement, and in at least 25% it is a presenting sign.
Nasal chondritis eventually develops in up to 70% of patients and may result in a saddle nose deformity. Symptoms include pain, stuffiness, crusting, rhinorrhea, epistaxis and compromise of olfaction. Nasal chondritis is typically less recurrent than the auricular chondritis, and the saddle nose deformity is more common in men .
Involvement of the cartilage of the respiratory tract (larynx, trachea, bronchi) and/or costochondral joints occurs in ~50% of patients and can be the most serious complication. Signs and symptoms include cough, hoarseness, choking, dyspnea, wheezing, or tenderness to palpation of the anterior neck in sites overlying the larynx or trachea. Complications include airway obstruction or collapse, flail chest, and secondary pulmonary infections.
Arthritis is common (50–80% of patients) and it is the presenting symptom in one-third of patients. An episodic, migratory, asymmetric, non-erosive oligo- or polyarticular arthritis can affect any joint, although the knees and MCP and proximal interphalangeal joints are those most commonly affected. There may also be involvement of the sternoclavicular and sternomanubrial joints.
Ocular inflammation develops in ~65% of patients. It can involve virtually any component of the eye, leading to conjunctivitis, scleritis, corneal ulcerations, uveitis, or optic neuritis .
A variety of reactive cutaneous lesions are seen in patients with relapsing polychondritis , including aphthae, small vessel vasculitis, annular urticarial plaques, livedo reticularis, (lymphocytic) Sweet syndrome, erythema elevatum diutinum, and erythema nodosum. None is pathognomonic and some may be coincidental or due to a coexisting disease. The presence of cutaneous small vessel vasculitis and/or aphthae is reported to increase the likelihood of an associated myelodysplastic syndrome (see Ch. 24 ). In addition, an overlap between relapsing polychondritis and Behçet disease, termed m outh a nd g enital ulcers and i nflamed c artilage (MAGIC) syndrome, has been reported .
Less common systemic features include audiovestibular damage, cardiovascular disease (aortitis, valvular dysfunction, pericarditis, conduction system abnormalities, myocarditis), renal dysfunction (glomerulonephritis, glomerulosclerosis, tubulointerstitial disease), and neurologic sequelae (cranial nerve palsies, vasculitis of the central or peripheral nervous system).
Pathology
Histopathologically, there is breakdown of the normal lacunar structure of the cartilage, with neutrophilic infiltrates initially, followed by lymphoplasmacytic infiltrates. In late stages there is replacement of cartilage by granulation tissue and fibrosis.
Differential Diagnosis
In the early inflammatory phase, the erythema and pain may be misdiagnosed as erysipelas or cellulitis. Infectious chondritis, traumatic chondritis, granulomatosis with polyangiitis (Wegener granulomatosis), small vessel vasculitis, and congenital syphilis may mimic the cartilage destruction seen in relapsing polychondritis ( Table 45.3 ).
| DIFFERENTIAL DIAGNOSIS OF NASAL DEFORMITY OR DESTRUCTION |
| Neoplastic disorders |
|
| Inflammatory disorders |
|
| Infectious disorders |
|
| Other etiologies |
|
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


