Abstract
This section focuses on genetic tumor predisposition syndromes that have characteristic mucocutaneous manifestations. Recognition of these skin findings facilitates diagnosis and surveillance for associated extracutaneous malignancies and endocrine abnormalities. Multiple endocrine neoplasia, Cowden syndrome, Gardner syndrome, and Muir–Torre syndrome are reviewed in detail.
This section provides an overview of enzyme deficiency diseases with skin findings that represent diagnostic clues. The diverse cutaneous features include discoloration, dermatitis, angiokeratomas, and sclerosis; adnexal manifestations such as hypohidrosis and alopecia may also be observed. Extracutaneous manifestations and current management strategies are highlighted. Conditions covered include defects in amino acid metabolism/transport, multiple carboxylase deficiency, lysosomal storage diseases, and mitochondrial disorders.
The classic premature aging syndromes are Hutchinson–Gilford progeria and Werner syndromes. However, additional rare disorders can be viewed as progeroid syndromes. Examples include MDPL ( m andibular hypoplasia, d eafness, p rogeroid features, and l ipodystrophy) syndrome and Néstor–Guillermo progeria syndrome. Understanding the underlying pathophysiology of progeria has led to insights into the group of disorders referred to as laminopathies as well as physiologic aging. This section also reviews disorders in which patients develop poikiloderma.
Ectodermal dysplasias are a diverse group of heritable disorders that share the primary feature of abnormalities in at least two of the major ectodermally derived structures – hair, teeth, nails, and sweat glands. Other ectodermal structures, such as mucous and sebaceous glands, are also frequently affected. The various forms of ectodermal dysplasia are distinguished based on the types of ectodermal abnormalities, presence of other cutaneous and extracutaneous manifestations, mode of inheritance, and underlying genetic defect. Ectodysplasin, WNT, and p63 signaling pathways have been implicated in the pathogenesis of both ectodermal dysplasias and inherited abnormalities limited to a single structure (e.g. hair, nails).
Keywords
multiple endocrine neoplasia, Cowden syndrome, PTEN hamartoma tumor syndrome, tricholemmomas, Gardner syndrome, familial adenomatous polyposis, Muir–Torre syndrome, Lynch syndrome, sebaceous neoplasms, alkaptonuria, angiokeratoma corporis diffusum, biotinidase deficiency, holocarboxylase synthetase deficiency, Fabry disease, fucosidosis, Gaucher disease, Hartnup disease, mitochondrial disorders, Niemann–Pick disease, phenylketonuria, progeroid syndromes, progeria, Werner syndrome, poikiloderma, nuclear envelopathies, laminopathies, lamin A, progerin, premature aging, ectodermal dysplasia, hypohidrotic ectodermal dysplasia, hidrotic ectodermal dysplasia, hypohidrosis, hypotrichosis, onychodystrophy, hypodontia, cleft lip/palate, ankyloblepharon, ectrodactyly
Introduction
In this chapter, a number of genodermatoses are discussed that are not covered in other chapters of this textbook ( Table 63.1 ).
| GENODERMATOSES |
|
|
Disorders characterized by cutaneous or mucosal tumors in conjunction with a variety of extracutaneous neoplasias, both benign and malignant, as well as endocrine abnormalities are the focus of the first section. It includes multiple endocrine neoplasia types 1, 2A, and 2B in addition to Cowden syndrome, Gardner syndrome, and Muir–Torre syndrome. This is followed by sections on: enzyme deficiencies; premature aging syndromes and poikilodermas; and ectodermal dysplasias.
Disorders Featuring Tumorigenesis
- Susan J. Bayliss
- Monique G. Kumar
Multiple Endocrine Neoplasia
Multiple endocrine neoplasia (MEN) refers to a group of disorders marked by the presence of neoplasia or hyperplasia in two or more endocrine organs, often in association with mucocutaneous findings. Three major MEN syndromes – termed types 1, 2A, and 2B – have been clinically and genetically defined and are described in Table 63.2 .
| TYPES OF MULTIPLE ENDOCRINE NEOPLASIA (MEN) | |||||||
|---|---|---|---|---|---|---|---|
| MEN type (synonyms) | Mode of inheritance | Gene locus | Gene | Protein product | Location and type of mutations | Clinical manifestations | |
| Systemic | Mucocutaneous | ||||||
| 1 (Wermer syndrome) | AD | 11q13 | MEN1 * [ MENIN ] | Menin (a nuclear protein) | Variable; truncating mutations in >70% |
|
|
| 2A (MEN type 2, Sipple syndrome) | AD | 10q11.2 | RET † | Tyrosine kinase receptor | Point mutations in the cysteine-rich extracellular domain, especially codon 634 ‡ |
| |
| 2B (MEN type 3, multiple mucosal neuroma syndrome) | AD | 10q11.2 | RET † | Tyrosine kinase receptor | >95% of patients have missense mutation (Met 918 Thr) in substrate-recognition pocket |
|
|
† Rearranged during transfection of proto-oncogene; mutations in the same gene can be responsible for Hirschsprung disease and familial medullary thyroid carcinoma.
‡ Induce dimerization and activation of receptor in the absence of its ligand.
§ Screen via measurement of serum calcitonin level after calcium or pentagastrin injection.
Mucocutaneous features are most prominent in MEN types 1 and 2B. In type 1, multiple facial angiofibromas and collagenomas are present in the majority of affected individuals ( Fig. 63.1 ), and lipomas are another common finding . Compared to those in tuberous sclerosis, facial angiofibromas in MEN 1 tend to be smaller and fewer in number, and they are more likely to arise on the upper lip and its vermilion border in addition to the nose and medial cheeks. Although melanoma has been reported in patients with MEN 1 and there is evidence that MEN1 expression is frequently lost in sporadic melanomas , it remains unclear whether individuals with this syndrome have an increased risk of melanoma.
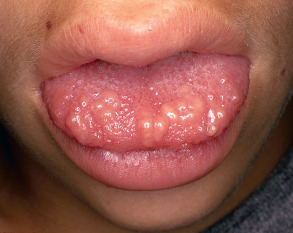
MEN 2B is notable for multiple mucosal neuromas, especially of the conjunctivae, lips, and anterior tongue ( Fig. 63.2 ). These lesions, which may be present at birth or develop during early childhood, are considered relatively specific for this diagnosis. However, mucosal neuromas are also occasionally observed in patients with the PTEN hamartoma tumor syndrome (see below). Cutaneous neuromas are rare in MEN 2B and when present are usually in a perinasal location.
Cowden Syndrome
▪ Multiple hamartoma syndrome ▪ Cowden disease ▪ PTEN hamartoma tumor syndrome spectrum
- ▪
Autosomal dominant disorder characterized by multiple hamartomatous tumors of ectodermal, mesodermal, and endodermal origin
- ▪
Mucocutaneous findings include tricholemmomas, acral keratoses, oral papillomas, neuromas, and sclerotic fibromas
- ▪
Patients have especially high risks of breast, thyroid, and endometrial carcinoma
- ▪
Caused by mutations in the PTEN gene
Introduction
PTEN hamartoma tumor syndrome (PHTS) refers to a spectrum of disorders with overlapping clinical features caused by mutations in the phosphatase and tensin homolog ( PTEN ) gene. Cowden syndrome (CS) represents the most common phenotype of PHTS and is characterized by multiple hamartomas of ectodermal, mesodermal, and endodermal origin. Macrocephaly and mucocutaneous lesions (e.g. tricholemmomas, acral keratoses, oral papillomas) are the most constant and characteristic features . Up to 85% of CS patients develop at least one malignant neoplasm, most often of the breast, thyroid, or endometrium .
History
In 1963, Lloyd and Dennis reported a multisystem disorder with characteristic mucocutaneous lesions and tumors of the breast, thyroid, and gastrointestinal tract . They named the disorder “Cowden disease” after their first patient.
Epidemiology
CS is probably more common than is reflected by the number of cases reported, because the clinical findings can be subtle. It is inherited as an autosomal dominant trait with variable expressivity; a female predominance has been noted in some series . Most of the reported patients have been Caucasian, and it is estimated to affect 1 in 200 000 individuals .
Pathogenesis
Heterozygous germline mutations in the tumor suppressor gene PTEN are found in most patients with CS as well as other conditions within the PHTS spectrum , which includes Bannayan–Riley–Ruvalcaba syndrome (BRRS), SOLAMEN syndrome ( s egmental o vergrowth, l ipomatosis, a rteriovenous m alformation and e pidermal n evus; see Ch. 62 ), and an autism/macrocephaly syndrome. Of note, there is significant overlap in the clinical findings of CS and BRRS ( Table 63.3 ). Identical mutations have been found in patients with CS and BRRS, and in some families both CS and BRRS phenotypes are observed.
| CLINICAL MANIFESTATIONS OF PTEN HAMARTOMA TUMOR SYNDROME AND SCREENING RECOMMENDATIONS | |||
|---|---|---|---|
| Clinical manifestations | |||
| Onset typically at birth or in childhood | Onset typically in adolescence or adulthood | Variable onset | |
| Mucocutaneous |
|
| |
| Craniofacial and skeletal |
| Scoliosis, kyphoscoliosis | |
| Thyroid |
| ||
| Breast |
| ||
| Gastrointestinal tract | Hepatic vascular anomalies | Adenocarcinoma (rare) |
|
| Genitourinary system |
| ||
| Central nervous system |
| Lhermitte–Duclos disease (LDD) – hamartomatous overgrowth of cerebellar ganglion cells that can lead to elevated intracranial pressure, ataxia and seizures ** |
|
| Ocular |
|
| |
| Muscular | Lipoid storage myopathy | ||
| Other | Neonatal macrosomia Persistent drooling | ||
| Screening recommendations | |||
| |||
* Typically multifocal lesions with fast-flow channels, intramuscular involvement, and associated ectopic fat; may have capillary, venous and/or lymphatic components.
† Includes tricholemmomas, lesions with histologic features of verrucae, and very rarely tumors of the follicular infundibulum.
‡ May represent epithelial hyperplasia, sclerotic fibromas, or glycogenic acanthosis.
§ Includes accelerated growth of the first metacarpal and first and second middle phalanges, syndactyly, and brachydactyly.
^ Can be hamartomatous, adenomatous, ganglioneuromatous or inflammatory.
** May occur as an isolated finding or in association with Cowden syndrome ; patients with adult-onset LDD usually have a mutation in PTEN .
The PTEN protein is a lipid and protein phosphatase that plays a role in cell-cycle regulation and apoptosis . In particular, PTEN downregulates the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway that promotes cellular proliferation and survival (see Fig. 55.3 ). As in other autosomal dominant disorders caused by a defective tumor suppressor gene, hamartomas and neoplasms typically result from a “second hit” somatic mutation that inactivates the remaining allele of the gene (loss of heterozygosity). In PHTS, this leads to excessive mTOR signaling in affected tissues, in particular those with physiologic proliferation, such as the epidermis, oral and gastrointestinal mucosa, and thyroid and breast epithelium. PTEN also has phosphatase-independent activities and nuclear functions independent of the PI3K/AKT/mTOR pathway .
Clinical Features
In 1995, the International Cowden Syndrome Consortium developed operational diagnostic criteria that have since been updated multiple times . The most recent criteria are outlined in Table 63.4 .
| REVISED CLINICAL DIAGNOSTIC CRITERIA FOR PTEN HAMARTOMA TUMOR SYNDROME (2013) |
| Major criteria |
|
| Minor criteria |
|
Operational diagnosis in an individual:
Operational diagnosis when a family member meets diagnostic criteria or has a PTEN mutation:
|
Mucocutaneous manifestations
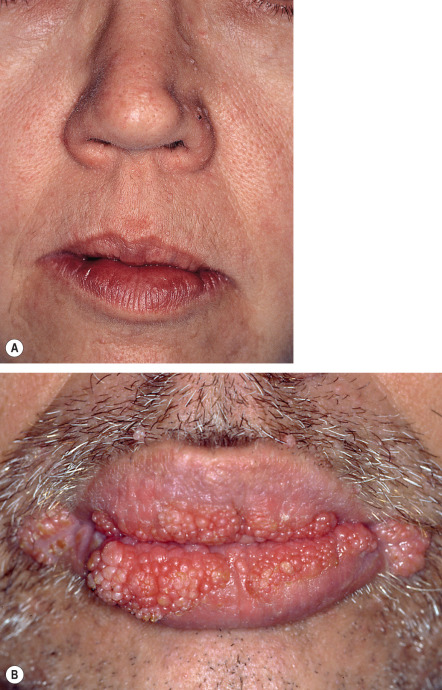
Cutaneous involvement occurs in >80% of patients with CS and often serves as an early sign of the disorder. Characteristic mucocutaneous lesions usually begin to appear during the second to third decades of life (average, 22 years; range, birth to 75 years). Facial tricholemmomas present as multiple skin-colored to yellowish-tan, verrucous, or keratotic papules that resemble common warts ( Fig. 63.3A ; see Ch. 111 ). Both tricholemmomas and nonspecific cutaneous papules show a predilection for the central face, and they may coalesce around facial orifices (especially the nostrils) and on the ears. Papules on the head and neck of CS patients may also represent inverted follicular keratoses or acrochordons.
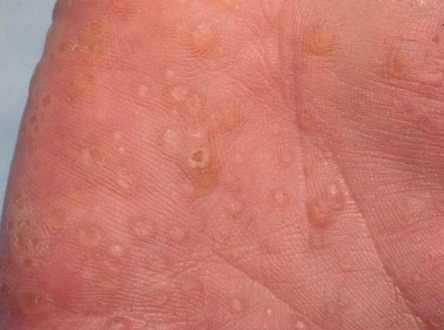
Multiple sclerotic fibromas of the skin are a relatively specific cutaneous marker of CS . These lesions present as sharply circumscribed, firm dermal papules that display a strikingly uniform storiform pattern of collagen bundles on histologic examination (see Ch. 116 ). Punctate palmoplantar keratoses, often translucent with a central depression, are present in more than half of patients ( Fig. 63.4 ). Keratotic papules can also be seen on the heels, the dorsal aspects of the hands and feet, and the extensor surfaces of the forearms and lower legs.
Other skin and soft tissue manifestations include lipomas (especially testicular lipomatosis), angiolipomas, and vascular anomalies. The vascular anomalies of CS and BRRS tend to be multifocal, fast-flow lesions with intramuscular involvement and associated ectopic fat . A distinctive “PTEN hamartoma of soft tissue” that presents as a subcutaneous and intramuscular mass with adipocytic, fibrous, and vascular (arterial, venous) components and sometimes lymphoid, bony, and neural elements has also been described . Lastly, recent series have suggested that CS patients may have a slightly increased risk of melanoma, with a lifetime incidence of ~5% .
Oral lesions of CS appear as 1–3 mm, asymptomatic, mucosa-colored papules that often lead to a cobblestone appearance ( Fig. 63.3B ). They have a predilection for the lips and tongue, but extensive involvement of the entire oral cavity can occur .
Extracutaneous manifestations
Multinodular goiter or thyroid adenomas occur in more than two-thirds of patients with CS, and the lifetime risk of thyroid carcinoma is estimated to be 10 to >30%. Fibrocystic disease and fibroadenomas of the breast affect three-quarters of female patients. Breast carcinoma develops in up to 85% of female patients with CS and is diagnosed at a mean age of approximately 40 years ; it has also been reported in affected men .
Multiple polyps may be found anywhere in the gastrointestinal tract but most frequently affect the colon (≥85% of patients). The lifetime risk of colon carcinoma is estimated to be ~10% . Benign ovarian cysts and uterine leiomyomas commonly develop and endometrial carcinoma occurs in as many as 20–30% of women with CS . The lifetime risk of renal carcinoma is estimated to be as high as ~30% .
Pathology
Histologically, tricholemmomas are characterized by a lobular proliferation of pale keratinocytes, often in association with a hair follicle. Basal keratinocytes are oriented to form a palisade, and hyperkeratosis can lead to a cutaneous horn. Demonstration of complete loss of PTEN expression within a tricholemmoma via immunohistochemical staining is suggestive that the patient has CS . Histologically, some facial papules in patients with CS may simply be squamous papillomas, while others show follicular infundibular hyperplasia. Oral lesions often represent fibromatous nodules with collagen fibers arranged in whorls. On biopsy, extrafacial and palmoplantar papules tend to be hyperkeratotic papillomas, and they may have changes reminiscent of verrucae or acrokeratosis verruciformis .
Differential Diagnosis
The clinical differential diagnosis of the oral papillomas of CS includes verrucae, focal epithelial hyperplasia (Heck disease), traumatic fibromas, mucosal neuromas of MEN 2B, papillomas of Goltz syndrome, and papular lesions of lipoid proteinosis. A combination of acral keratoses (including palmoplantar) plus oral and facial papules can also be seen in Darier disease, whereas multiple facial angiofibromas and gingival fibromas are characteristic of tuberous sclerosis. These disorders can usually be differentiated from CS by other characteristic features as well as histologic evaluation.
The clinical differential diagnosis of multiple facial tricholemmomas may include angiofibromas (as seen in tuberous sclerosis or MEN 1), trichoepitheliomas, and fibrofolliculomas (Birt–Hogg–Dubé syndrome); in addition to their histologic features, the tendency of tricholemmomas to have a verrucous appearance clinically may help to distinguish them from these lesions. Common warts, seborrheic keratoses, basaloid follicular hamartoma syndrome, and epidermodysplasia verruciformis may represent additional diagnostic possibilities. Less commonly, multiple syringomas or the basal cell nevus syndrome might be considered (see Fig. 111.5 ).
Germline mutations in the SDHB/C/D genes, which encode subunits of mitochondrial s uccinate d e h ydrogenase, have been associated with an autosomal dominant CS-like disorder characterized by a predisposition to the development of breast, thyroid, and renal carcinomas; to date, cutaneous manifestations have not been described . Predisposition to the same malignancies as well as endometrial carcinoma has been associated with germline epigenetic alteration in the KLLN gene, which encodes a p53-regulated DNA replication inhibitor . One study found that ~10% of patients with a CS-like disorder but no PTEN or SDHB/C/D mutations had a germline mutation in PIK3CA or AKT1 , which encode proteins that activate mTOR signaling (see Fig. 55.3 ); cutaneous findings such as tricholemmomas and lipomas were noted in a few affected individuals.
Treatment
Although the recognition of CS and other forms of PHTS is based upon clinical and histologic evaluation, genetic testing can help to confirm the diagnosis and facilitate identification of affected family members. Due to the high risk of malignancy, cancer surveillance is a critical part of PHTS management (see Table 63.3 ).
Agents that downregulate the mTOR pathway, such as sirolimus (rapamycin), represent promising therapeutic approaches for PHTS that are currently under investigation, with reports of improvement of infiltrative vascular anomalies and lipomatosis following treatment . The facial papules of CS respond variably to topical 5-fluorouracil, oral isotretinoin, curettage, laser ablation, or surgical excision .
Gardner Syndrome
▪ Familial polyposis of the colon
- ▪
Autosomal dominant disorder characterized by premalignant intestinal polyposis and adenocarcinoma of the gastrointestinal tract
- ▪
Extraintestinal manifestations include epidermoid cysts, osteomas, desmoid tumors and fibrous tumors (skin, mesentery, retroperitoneum)
- ▪
Congenital hyperpigmentation of the retinal pigment epithelium (CHRPE) is an early sign
- ▪
Caused by mutations in the APC (adenomatous polyposis coli) gene
Introduction
The main features of Gardner syndrome (GS) are premalignant intestinal polyposis, epidermoid cysts, osteomas, and desmoid or fibrous tumors of the skin and other sites. It is considered to represent a phenotypic variant of the familial adenomatous polyposis syndrome with prominent extraintestinal involvement .
History
In 1953, Gardner and Stephens described a large family in which many individuals had multiple cutaneous lesions, osteomatosis and fatal bowel cancer. The syndrome now bears Gardner’s name.
Epidemiology
The incidence of GS is approximately 1 in 8000 to 1 in 16 000 births. It is inherited as an autosomal dominant trait with high penetrance and variable expressivity, affecting men and women equally.
Pathogenesis
GS is caused by heterozygous germline mutations in the adenomatous polyposis coli ( APC ) tumor suppressor gene. The APC gene encodes a protein that downregulates the Wnt/β-catenin signaling pathway, which has important functions in cellular proliferation, differentiation and adhesion (see Fig. 55.6 ).
Clinical Features
Cutaneous and soft tissue manifestations
Skin lesions and bony abnormalities appear during childhood and adolescence, frequently preceding the onset of polyposis. Cutaneous epidermoid cysts affect ~30–50% of patients with GS and are often multiple. These lesions are commonly found on the head and neck, may be present at birth, and tend to increase in size and number and then stabilize. Multiple familial pilomatricomas have also been reported as a presentation of GS .
Desmoid tumors are non-encapsulated, non-metastasizing, locally aggressive benign tumors that occur in 10–20% of patients with GS. There is a marked female predominance (70% to 85%). Desmoid tumors may occur spontaneously or at incision sites, arising from musculo-aponeurotic soft tissues. They commonly develop after colectomy and most often arise within the abdominal wall or intra-abdominally, with the latter causing greater morbidity via obstruction of the small bowel and/or ureters. Extra-abdominal lesions affecting the shoulder girdle, chest wall, or inguinal region can also occur. Although cytologically benign, desmoid tumors can be locally aggressive and may lead to substantial morbidity and even mortality. Hereditary desmoid disease characterized by multiple desmoid tumors, often at unusual sites, in patients with few or no colonic polyps has been described in association with APC mutations .
Fibromas may occur in the skin, subcutaneous tissues, mesentery, or retroperitoneum. The “Gardner fibroma” often arises in the first decade of life, favors the back and paraspinal region, and can serve as a desmoid precursor. Lipomas, leiomyomas, trichoepitheliomas, neurofibromas, and ovarian cysts are observed less commonly.
Other extraintestinal manifestations
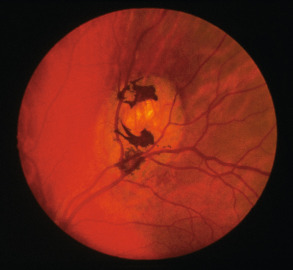
Congenital hypertrophy of the retinal pigment epithelium (CHRPE) is evident in ~75% of patients with GS and can be an early sign of the disorder ( Fig. 63.5 ). Because CHRPE is present at birth and easily detected by ophthalmologic examination, it is a useful finding in patients who do not yet have other manifestations. While unilateral solitary CHRPE may occur in unaffected individuals, multiple and bilateral lesions represent a sensitive and specific marker for GS. There are rare reports of adenocarcinoma arising from CHRPE.
Osteomas occur in ~80% of patients and favor the mandible and maxilla, although they can develop in other bones of the skull and in long bones. Seen in children as young as 8 years of age, osteomas are painless and may grow large enough to be clinically obvious or they may be detectable only by radiographic studies. Other skeletal lesions include exostoses, endostoses, and cortical thickening of the long bones. Dental anomalies are seen in ~20% of patients with GS and include supernumerary teeth, odontomas, unerupted teeth, and multiple caries.
Patients with GS are at increased risk for the development of other tumors, including papillary carcinoma of the thyroid (particularly in women), hepatoblastoma (usually affecting children less than 3 years of age), pancreatic and biliary tract carcinomas, adrenal adenomas (or occasionally carcinomas), and various sarcomas (e.g. fibrosarcoma, osteosarcoma). The association of brain tumors (most often medulloblastomas) with adenomatous polyposis in patients with heterozygous APC mutations has historically been referred to as Turcot syndrome type 2. The latter is distinct from the constitutional mismatch repair deficiency syndrome (previously referred to as Turcot syndrome type 1) that presents during childhood with brain tumors (primarily glioblastomas), colonic polyposis, hematologic malignancies, and multiple café-au-lait macules due to biallelic mutations in mismatch repair genes (see below and Table 61.4 ) .
Gastrointestinal manifestations
In adults, the diagnosis of GS is commonly made when the patient presents with gastrointestinal bleeding secondary to adenomatous polyps. Other findings may include anemia, abdominal pain, diarrhea or constipation, and weight loss. Premalignant adenomatous polyps most commonly occur in the colon but are also found in the small intestine and stomach in over half of patients. Polyps are typically <1 cm in diameter and are rarely seen before the age of 10 years. By the age of 20 years, 50% of patients with GS have demonstrable polyps. Without surgical intervention, colorectal carcinoma is inevitable; it usually develops before age 40 years and arises by early adolescence in approximately 5% of affected individuals . Carcinomas of the small intestine (especially the duodenum) can also occur .
Pathology
Histologically, intestinal polyps show adenomatous hyperplasia of the mucosa. Cutaneous cysts in patients with GS usually demonstrate the same microscopic changes as ordinary epidermoid cysts, with a lining of keratinizing epithelium that includes a granular layer. Pilomatricoma-like changes, with characteristic basophilic and shadow cell populations, have also been noted in the cysts of GS.
Differential Diagnosis
Epidermoid cysts usually occur in the absence of a particular syndrome, but multiple lesions should prompt a thorough family history and evaluation for other findings of GS (e.g. osteomas, CHRPE). In Peutz–Jeghers syndrome, mucocutaneous lentigines (intraoral, perioral, and acral) are seen and the gastrointestinal polyps are hamartomatous rather than adenomatous. Hamartomatous gastrointestinal polyps are also found in juvenile polyposis syndromes (which may be associated with a hereditary hemorrhagic telangiectasia phenotype; see Table 104.2 ) and PTEN hamartoma tumor syndrome. Additional manifestations such as osteomas, CHRPE, and desmoid tumors distinguish GS from these conditions. Of note, mutations in the MUTYH base excision repair gene can cause an autosomal recessive form of colorectal adenomatous polyposis, with osteomas, CHRPE, desmoid tumors, pilomatricomas, and sebaceous neoplasms (see below) having been described in affected individuals .
Treatment
Genetic counseling is imperative, and family members should be evaluated for extraintestinal and intestinal signs of the disorder. Molecular testing for APC mutations is commercially available.
Because of the inevitable development of colorectal carcinoma, prophylactic colectomy is recommended when polyp formation becomes evident, and it is usually performed between 15 and 25 years of age. Initially, children with GS should be monitored with annual flexible sigmoidoscopy or colonoscopy beginning at 10–12 years of age. Once polyps are detected, patients should undergo annual colonoscopy until a colectomy is done. Upper gastrointestinal endoscopy is recommended every 1–3 years (depending on polyp burden), with careful inspection of the ampulla of Vater because of the increased risk of cancer in this area. Barium studies or (preferably) capsule endoscopy can be utilized periodically to evaluate the remainder of the small bowel. Because of the increased risk of hepatoblastoma, some experts recommend annual abdominal ultrasounds and measurement of serum α-fetoprotein levels during the first 5 years of life.
Symptomatic epidermoid cysts can be surgically excised. Desmoid tumors are also treated by surgical excision, although there tends to be a high recurrence rate. Preoperative MRI of the abdomen can demonstrate the origin of the tumor and the extent of involvement. If wide surgical excision fails, radiotherapy, hormonal therapy (e.g. tamoxifen, raloxifene), chemotherapy, or a combination thereof can be tried. The osteomas of GS may or may not be amenable to surgical intervention. Medications that affect prostaglandin metabolism, such as nonsteroidal anti-inflammatory drugs (NSAIDs), may reduce the risk of colorectal cancer and desmoids .
Muir–Torre Syndrome
▪ Cutaneous sebaceous neoplasms and keratoacanthomas, with gastrointestinal and other carcinomas
- ▪
An autosomal dominant disorder associated most commonly with adenocarcinoma of the colon, followed by genitourinary carcinomas
- ▪
The cutaneous manifestations are sebaceous neoplasms and keratoacanthomas
- ▪
Caused by mutations in three DNA mismatch repair genes ( MSH2 , MLH1 , MSH6 )
Introduction
Muir–Torre syndrome (MTS) is an uncommon autosomal dominant genodermatosis that is characterized by sebaceous neoplasms (adenoma, sebaceoma, or carcinoma; see Ch. 111 ), keratoacanthomas, and internal malignancies. It is considered to represent a subtype of Lynch syndrome, which is also known as hereditary non-polyposis colorectal cancer (HNPCC) syndrome .
History
Muir in 1967 and Torre in 1968 independently reported the syndrome that bears their names. Their two patients had malignancies in several organs plus either keratoacanthoma-like lesions or multiple tumors of sebaceous origin.
Epidemiology
The male : female ratio in MTS is 3 : 2, and affected individuals present with cutaneous manifestations at a mean age of 55 years. The prevalence of Lynch syndrome in the general population is estimated to be ~1 in 500. In a study of Lynch syndrome patients, MTS was observed in 28% (14/50) of families and 9% (14/152) of individuals with a confirmed germline mismatch repair gene mutation .
Pathogenesis
MTS is caused by heterozygous germline mutations in one of several DNA mismatch repair genes, including MSH2 (70–90% of MTS cases), MLH1 , and MSH6 . In patients with MTS, somatic inactivation of the other allele of the affected gene (loss of heterozygosity) results in microsatellite instability, i.e. increased variability in the lengths of repetitive “microsatellite” sequences in the genome, as well as mutations that lead to tumor formation .
Clinical Features
Sebaceous tumors can present prior to (~30%), concurrently with (~10%), or after (~60%) the diagnosis of a visceral malignancy. The skin tumors sometimes appear decades before or after a visceral malignancy develops. In general, sebaceous tumors favor the head and neck region ( Fig. 63.6 ), and sebaceous carcinomas tend to affect the periocular area. However, in patients with MTS, these neoplasms often occur in other locations (trunk > extremities) . Sebaceous adenomas are the most common tumor type, and they present as yellow papules or nodules numbering from one to hundreds . Keratoacanthomas, which are keratotic skin-colored to erythematous papules or nodules that arise quickly, are seen in ~25% of patients with MTS and may exhibit sebaceous differentiation.
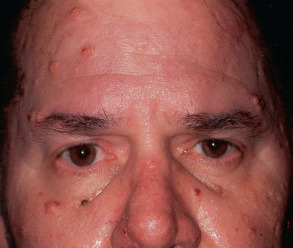
The most frequently observed malignancies are colorectal (61%) and genitourinary (22%; endometrium, ovary, urinary tract) carcinomas . Colorectal carcinoma in MTS most often involves the proximal colon and develops at a median age of 50 years, about one decade before colorectal carcinoma typically occurs in the general population. In addition, MTS patients can develop breast carcinomas (6%); hematologic malignancies (11%); head and neck cancers (5%); and neoplasms of the small intestine (3%), stomach, or pancreas .
The diagnosis of MTS is based on the presence of at least one sebaceous tumor (or keratoacanthoma with sebaceous differentiation) and a visceral malignancy in the absence of any known predisposing factor. In the absence of a sebaceous tumor, the diagnosis can be made in a patient with multiple keratoacanthomas, multiple visceral malignancies, and a family history of MTS. Findings that suggest a higher or lower likelihood of MTS in patients with sebaceous neoplasms are summarized in Table 63.5 . Testing for a mismatch repair gene mutation is helpful for confirming the diagnosis and facilitating early identification and surveillance of affected family members.
| RISK ASSESSMENT FOR MUIR–TORRE SYNDROME (MTS) | |
|---|---|
| Higher likelihood of MTS | Lower likelihood of MTS |
| Clinicopathologic characteristics of skin lesions | |
|
|
| Characteristics of patient and family history | |
|
|
| Additional analysis of sebaceous neoplasms | |
| |
* Especially if MSI-high histology – presence of tumor-infiltrating lymphocytes, Crohn disease-like lymphocytic reaction, medullary growth pattern, or signet ring/mucinous differentiation.
‡ MSH2 and MSH6 proteins form a heterodimer, so lack of one can lead to the absence of the other; likewise, PMS2 is unstable without a functional MLH1 protein.
§ Testing can be performed on paraffin-embedded tissue; diagnostic yield may be higher for colonic neoplasms.
Pathology
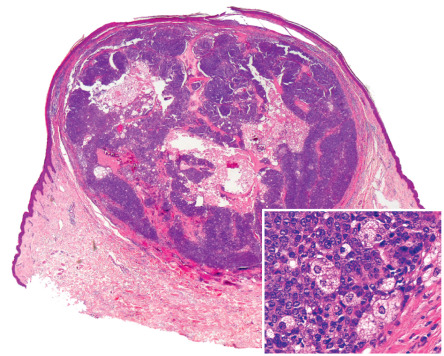
Skin tumors arising as part of MTS fall into two general categories: sebaceous neoplasms and keratoacanthomas. Sebaceous adenomas (with a predominance of mature sebocytes), sebaceomas (vertically oriented with a predominance of basaloid seboblastic cells), so-called sebaceous epitheliomas (inconsistently defined term used for various lesions with seboblastic differentiation), and sebaceous carcinomas may occur (see Ch. 111 ). A type of sebaceous adenoma that may be specific to this syndrome has been called a seboacanthoma; it has the architecture of a keratoacanthoma but contains well-differentiated sebaceous lobules ( Fig. 63.7 ) . In addition, cystic sebaceous tumors, which are well-circumscribed, deep sebaceous lesions with a cystic growth pattern, have been identified in some MTS patients .
Immunohistochemical (IHC) staining for MSH2, MSH6, MLH1, and PMS2 can be a valuable screening tool for MTS-associated tumors, which frequently show a lack of expression of one or more of these mismatch repair proteins (see Table 63.5 ) . However, abnormal IHC staining is sometimes seen in sporadic sebaceous neoplasms, especially in immunocompromised patients; conversely, IHC staining is occasionally normal in lesions from patients with a mismatch repair gene mutation (see Ch. 111 ) . PCR-based assays can also be used to demonstrate microsatellite instability (MSI) in tissue from MTS-associated neoplasms. Confirmatory testing for a germline mismatch repair gene mutation should be considered in patients with abnormal lesional IHC staining for these proteins, evidence of MSI, or a strong clinical suspicion of MTS.
Differential Diagnosis
Sebaceous hyperplasia is a common finding in normal individuals and sebaceous adenomas and carcinomas can occur in patients without MTS. Sebaceous adenomas and carcinomas as well as colorectal carcinomas and other malignancies have been described in patients with autosomal recessive colorectal adenomatous polyposis caused by mutations in the MUTYH base excision repair gene . Multiple keratoacanthomas may occur as an isolated finding (i.e. without visceral malignancy), especially in patients with the Ferguson–Smith (multiple self-healing) or Grzybowski (generalized eruptive) types of keratoacanthomas. However, these patients should be evaluated to exclude the possibility of MTS.
Treatment
Patients with an internal malignancy require appropriate oncologic referral and management. Recommendations for surveillance of high-risk individuals are outlined in Table 63.6 . Long-term aspirin therapy may decrease the risk of colorectal carcinoma in patients with Lynch syndrome .
| SURVEILLANCE GUIDELINES FOR PATIENTS WITH LYNCH SYNDROME, INCLUDING MUIR–TORRE SYNDROME | ||
|---|---|---|
| Test | When to begin | Frequency |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
* Hysterectomy and bilateral salpingo-oophorectomy are recommended when childbearing is completed or at age 40 years.
Treatment options for the benign sebaceous tumors include curettage, excision, cryosurgery and radiotherapy. Mohs micrographic surgery is recommended for sebaceous carcinomas on the face. Oral retinoids with or without interferon-α may be useful in the prevention of new sebaceous neoplasms.
Enzyme Deficiency Diseases
- Ángela Hernández-Martín
- Barbara K. Burton
Alkaptonuria
▪ Ochronosis
- ▪
An autosomal recessive disorder due to deficiency of homogentisate 1,2-dioxygenase
- ▪
Clinical signs include pigmentation of cartilage (e.g. the helices), sclerae, and skin (e.g. the axillae)
- ▪
Extracutaneous findings include urine that darkens on standing, arthritis, and valvular heart disease
The cardinal clinical features of alkaptonuria include urine that turns dark on standing, pigmentation of cartilage and other connective tissues, and arthritis ( Fig. 63.8 ). The arthritis often presents in the third or fourth decade of life with chronic back pain and stiffness, and radiographs show flattened and calcified intervertebral discs. Later involvement of large peripheral joints can resemble rheumatoid arthritis clinically but osteoarthritis radiographically. Renal complications include an increased incidence of calculi and occasionally kidney failure. Aortic stenosis is common by the sixth or seventh decade of life and coronary artery calcifications may also develop . Life expectancy is normal.
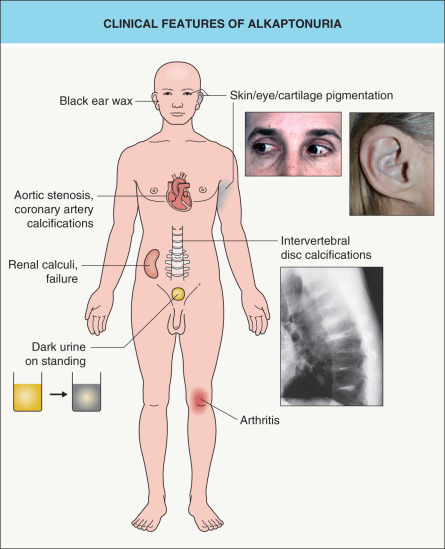
The dermatologic features of alkaptonuria are rarely noted prior to 10–15 years of age and typically become apparent during adulthood, often in the fourth decade of life. Axillary skin may be one of the initial sites to develop discoloration, which can range in color from blue to yellow to brown. The classic blue–grey pigmentation associated with this disorder is often first noted in the helices of the ear and sclerae (see Fig. 63.8 ). Later, it may affect the entire face as well as the palmar and plantar surfaces. Features that may present early in life include brownish discoloration of diapers (due to the dark urine) and cerumen that is brown to black in color.
Diagnosis of alkaptonuria can be confirmed by urine organic acid analysis. Management includes physical therapy and pain control as needed. Beginning at age 40 years, periodic cardiac assessment with echocardiography is recommended . Administration of oral nitisinone, an inhibitor of HGA production, at a dose of 2 mg daily reduces urinary HGA excretion by >95%, although the clinical benefits remain to be determined; a low-protein diet in conjunction with this medication may help to prevent excessively high plasma tyrosine levels .
Biotinidase and Holocarboxylase Synthetase Deficiencies
▪ Late- and early-onset forms of multiple carboxylase deficiency
- ▪
As biotin is an essential cofactor for carboxylases, there is overlap in these two autosomal recessive disorders
- ▪
Cutaneous findings include alopecia and periorificial erythema and erosions, features of “nutritional dermatitis”
- ▪
Metabolic acidosis occurs in both disorders
- ▪
Biotinidase deficiency is associated with developmental delay, hearing loss, and seizures beginning during infancy or childhood, while patients with holocarboxylase synthetase deficiency classically develop encephalopathy and hypotonia within the first 3 months of life
Biotinidase deficiency , a pan-ethnic disorder with an incidence of approximately 1 in 60 000 births, represents a defect in biotin recycling. Patients with this disorder become functionally biotin-deficient under conditions of normal dietary biotin intake. Since biotin is an essential cofactor for the activity of carboxylases, multiple carboxylase deficiency ensues. The clinical manifestations of the disorder, although entirely preventable with the administration of 5–20 mg of biotin daily, are highly variable and may go unrecognized. This has led to the inclusion of biotinidase deficiency in the newborn screening panel of a number of countries, including the US . Both cutaneous and extracutaneous manifestations usually develop between 3 months and 2 years of age but may be delayed until later childhood. Extracutaneous features include developmental delay, hearing loss, seizures, conjunctivitis, optic atrophy, myelopathy, and metabolic acidosis. Abnormalities on urine organic acid analysis are not consistently present. The diagnosis is established by an assay of biotinidase activity in blood; prenatal diagnosis is possible via enzymatic and molecular analyses.
Holocarboxylase synthetase deficiency is significantly less common than biotinidase deficiency. It represents a defect in biotinylation of the apocarboxylases, leading to a failure in synthesis of intact functional holocarboxylases. Patients typically present within the first 3 months of life with symptoms of a metabolic encephalopathy, including poor feeding, lethargy, respiratory distress, and hypotonia. Metabolic acidosis, hyperammonemia, and organic aciduria are often observed. Definitive diagnosis is established by assays of carboxylases in leukocytes or cultured skin fibroblasts; prenatal diagnosis is also possible via enzymatic and molecular analyses. Most patients exhibit significant improvement when treated with 10 mg of biotin daily.
Fabry Disease
▪ Anderson–Fabry disease
- ▪
X-linked disorder due to a deficiency of α-galactosidase A
- ▪
Characteristic mucocutaneous finding is multiple angiokeratomas
- ▪
Additional features include pain and paresthesias of the extremities, hypohidrosis, and renal and coronary insufficiency
Fabry disease classically presents during childhood or adolescence with the cardinal clinical features of acral pain, acral paresthesias, hypohidrosis, and angiokeratomas ( Fig. 63.9 ). Characteristic whorled corneal opacities (“cornea verticillata”) are also evident in ~75% of both male hemizygotes and female heterozygotes by childhood or adolescence; posterior lenticular opacities and tortuous retinal or conjunctival vessels may also be observed. However, these early findings can be subtle or absent in atypical variants of Fabry disease with delayed onset.
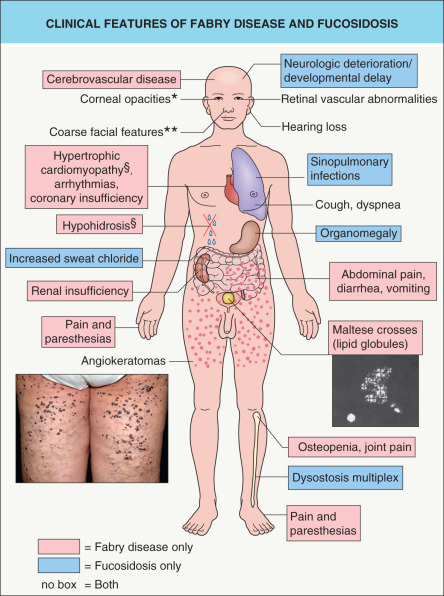
With increasing age, progressive renal insufficiency develops, leading to the need for dialysis or transplantation by the fourth or fifth decade of life in most male patients. Cardiac sequelae such as hypertrophic cardiomyopathy, arrhythmias, valvular abnormalities, and coronary insufficiency occur in most affected men by middle age and represent the most common cause of premature death in both men and women with Fabry disease. Manifestations in other organ systems can include: transient ischemic attacks and strokes; abdominal pain, diarrhea, and vomiting (especially in children); obstructive lung disease; joint pain and osteopenia; and hearing loss . Characteristic facial features such as periorbital fullness, bushy eyebrows, a broad nasal bridge, thick lips, prognathism, and prominent earlobes have been noted in men with Fabry disease.
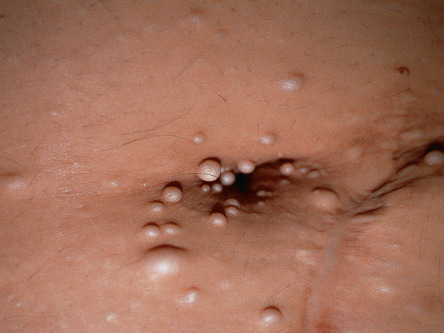
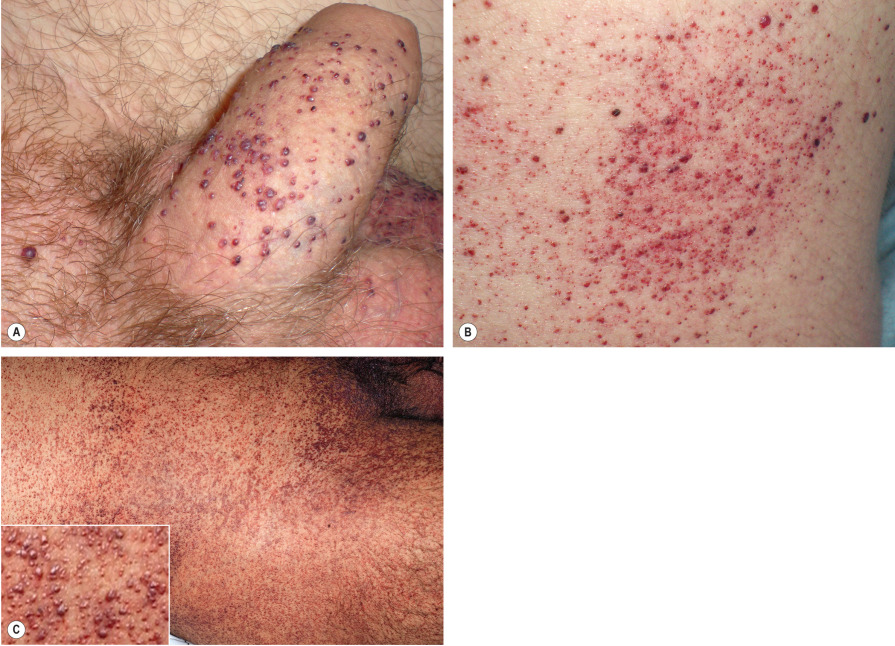
Angiokeratomas are one of the earliest signs of Fabry disease, typically beginning to appear during childhood or adolescence . They develop in most male patients and ~30% of female heterozygotes. Angiokeratomas present as punctate, dark-red to blue–black macules or papules ( Fig. 63.10 ). The lesions do not blanch with pressure and may become slightly keratotic as they enlarge. The angiokeratomas of Fabry disease, referred to as angiokeratoma corporis diffusum, tend to be concentrated between the umbilicus and the knees but can develop at any cutaneous site (see Fig. 63.10 ); oral and conjunctival lesions are also seen. Less frequent vascular findings include linear telangiectasias favoring the face, lips, and oral mucosa; episodic acral vasospasm resembling Raynaud phenomenon, typically accompanied by pain and paresthesias; and peripheral edema and lymphedema . Hypohidrosis is an early and almost constant feature of Fabry disease that can result in heat intolerance. Decreased body hair has also been described.
Histologically, angiokeratomas are composed of dilated capillaries in the uppermost dermis, partially enclosed by elongated rete ridges (see Ch. 114 ); hyperkeratosis may be seen in older lesions. In the skin, there is evidence of lipid storage in endothelial cells, pericytes, arteriolar smooth muscle, and arrector pili muscle. This can be detected by a lipid stain such as Sudan black B, or highlighted by periodic acid Schiff (PAS) staining. Lipid accumulation may also be observed in sweat gland epithelium (see Ch. 39 ) and perineural cells. Ultrastructural examination demonstrates characteristic cytoplasmic inclusions in all of these cell types.
The diagnosis of Fabry disease can be established by demonstration of deficient α-galactosidase A activity in plasma, leukocytes, or cultures of amniocytes or chorionic villi obtained from prenatal testing. However, enzyme activity is within the normal range in up to a third of female heterozygotes. Analysis of the GLA gene is clinically available and can be helpful in: (1) determining disease status in female patients; (2) predicting disease severity in affected male patients; and (3) establishing a prenatal/preimplantation diagnosis. Polarizing microscopy of urine from Fabry disease patients reveals birefringent lipid globules (“Maltese crosses”) (see Fig. 63.9 ). Although most commonly seen in association with Fabry disease, angiokeratoma corporis diffusum can also be observed in several other lysosomal storage disorders, as outlined in Table 63.7 . Scrotal and vulvar angiokeratomas are also commonly found in older adults (see Ch. 114 ).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


