Abstract
The non-infectious granulomatous diseases discussed in this chapter are unified by similar histologic findings despite having disparate, often unknown, causes. They are defined by their cutaneous inflammatory infiltrate, consisting of histiocytes, macrophages, giant cells, and granuloma formation. The etiopathogenesis, risk factors, disease associations, and in many cases therapeutic ladders for evaluating and managing patients with these uncommon diseases are in many instances based on case reports, small case series, retrospective studies, and expert opinion. Robust high-quality data is often lacking.
Keywords
sarcoidosis, granulomatous, non-infectious granulomatous, granuloma annulare, multinucleated giant cells, necrobiosis lipoidica, cutaneous Crohn disease, annular elastolytic giant cell granuloma, interstitial granulomatous dermatitis, palisaded neutrophilic and granulomatous dermatitis
Introduction
The granulomatous diseases discussed in this chapter may be defined as having an inflammatory cutaneous infiltrate in which histiocytes are the preponderant inflammatory cells. While cutaneous sarcoidosis is the prototype of non-infectious (sterile) granulomatous dermatitides, there are a number of other entities in this group ( Fig. 93.1 & Table 93.1 ). Additional non-infectious granulomatous disorders are covered in other chapters, including rheumatoid nodules in Chapter 45 , granulomatous cheilitis in Chapter 72 , foreign body granulomas in Chapter 94 , and primary immunodeficiency-associated granulomatous dermatitis in Chapter 60 .
| CLINICAL FEATURES OF THE MAJOR NON-INFECTIOUS GRANULOMATOUS DERMATITIDES | ||||||
|---|---|---|---|---|---|---|
| Sarcoidosis * | Classic granuloma annulare † | AEGCG | Necrobiosis lipoidica | Cutaneous Crohn disease | Rheumatoid nodule | |
| Average age (years) | 25–35, 45–65 | <30 | 50–70 | 30 | 35 | 40–50 |
| Sex predilection | Female | Female | Female | Female | Female | Male ‡ |
| Racial/ethnic predilection in US | African-American | None | Caucasian | None | Ashkenazi Jews | None |
| Sites | Symmetric on face, neck, upper trunk, extremities | Hands, feet, extensor aspects of extremities | Face, neck, forearms (sites of chronic sun exposure) | Anterior and lateral aspects of shins | Anogenital region, buttocks, lower > upper extremities | Juxta-articular areas, especially elbows and hands |
| Appearance | Protean; most commonly red– brown or violaceous papules and plaques | Papules coalescing into annular plaques | Annular plaques | Plaques with elevated borders, telangiectasias centrally | Dusky erythema, lymphedema, ulceration, vegetating plaques | Skin-colored, firm, mobile subcutaneous nodules |
| Size of lesions | 0.2 to >5 cm | 1–3 mm papules, annular plaques usually <6 cm | 2 to >10 cm | 3 to >10 cm | Variable | 1–3 cm |
| No. of lesions | Variable | 1–10 | 1–10 | 1–10 | 1–5 | 1–10 |
| Associations | Systemic manifestations of sarcoidosis (see Table 93.3 ); can be drug-induced (e.g. IFN, TNF inhibitors) or reaction pattern to underlying lymphoma | Possible diabetes mellitus, thyroid disease, or hyperlipidemia; rare reports of HIV infection, malignancy | Actinic damage | Diabetes mellitus | Intestinal Crohn disease | Rheumatoid arthritis |
| Special clinical characteristics | Develop within sites of trauma including scars and tattoos | Central hyperpigmentation | Central atrophy and hypopigmentation | Yellow–brown atrophic centers, ulceration | Draining sinuses and fistulas | Occasional ulceration, especially at sites of trauma |
* Clinical variants include lupus pernio and subcutaneous (Darier–Roussy), psoriasiform, ichthyosiform, angiolupoid, and ulcerative sarcoidosis.
† Clinical variants include generalized/disseminated, micropapular, nodular, perforating, subcutaneous, and patch granuloma annulare.
‡ Although rheumatoid arthritis has a female : male ratio of 2–3 : 1.
Sarcoidosis
▪ Löfgren syndrome – erythema nodosum, hilar adenopathy, fever, arthritis ▪ Heerfordt syndrome – parotid gland enlargement, uveitis, fever, cranial nerve palsy ▪ Darier–Roussy disease – subcutaneous sarcoidosis
- ▪
A systemic granulomatous disorder of unknown origin that most commonly involves the lungs
- ▪
Cutaneous manifestations of sarcoidosis are seen in up to one-third of patients and may be the first clinical sign of the disease
- ▪
Red–brown to violaceous papules and plaques appear most often on the face, in particular the nose, neck, upper back and extremities, as well as within scars and tattoos
- ▪
Erythema nodosum is a nonspecific inflammatory skin finding that may be associated with an acute form of sarcoidosis which tends to remit
- ▪
Histologically, sarcoidosis is characterized by non-caseating epithelioid granulomas, usually with a sparse or absent surrounding lymphocytic inflammation (i.e. “naked” granulomas)
History
Sarcoidosis was described initially by Sir Jonathan Hutchinson in 1875 and cutaneous sarcoidosis (lupus pernio) by Besnier in 1889. Ten years later, Caesar Boeck coined the term “multiple benign sarkoid”, noting that the lesions resembled benign sarcomas.
Epidemiology
Sarcoidosis, which occurs in patients of all races and ages as well as both sexes, is characterized by a bimodal age distribution, with peaks between 25 and 35 years and then between 45 and 65 years in women. In the US, there is an increased incidence of sarcoidosis in African-Americans, ranging from 35 to 64 per 100 000. Middle-aged African-American women have the highest incidence (107 per 100 000), with African-American women having a lifetime risk of developing the disease of 2.7% . In comparison, the incidence in Caucasians in the US ranges from 10 to 14 per 100 000. Sarcoidosis in African-Americans also tends to be more chronic and severe with a higher mortality rate than in other populations . Worldwide, the incidence of sarcoidosis is highest in Sweden (64 per 100 000) and the UK (20 per 100 000) and lowest in Spain and Japan (both 1.4 per 100 000). A greater number of patients with new-onset sarcoidosis are reported in the winter and spring .
Pathogenesis
Immune mechanisms
Sarcoidosis is a multisystem granulomatous disease characterized by hyperactivity of the cell-mediated immune system. Patients with a genetic susceptibility are exposed to a triggering antigen, leading to activation of macrophages and T cells, with subsequent granuloma formation. While classically considered a Th1-predominant immune response, the inflammatory cascade in sarcoidosis likely spans multiple pathways, including the innate immune system (via activation of pattern recognition receptors such as Toll-like receptors or NOD-like receptors) and potentially the Th17 arm of the immune system . Specifically, upregulation of CD4 + T helper cells of the Th1 subtype occurs following antigen presentation by monocytes bearing MHC class II molecules, which initiates formation of epithelioid granulomas in a variety of tissue types . In the lung, an oligoclonal α/β T-cell population has been described, suggesting that antigenic triggers of sarcoidosis favor a progressive accumulation and activation of specific T-cell clones .
Increased production of Th1 cytokines, including interleukin (IL)-2, IL-12, IL-18 and interferon (IFN)-γ, as well as release of tumor necrosis factor (TNF)-α by macrophages and some CD8 + T cells, leads to persistent Th1 activity and persistent IFN-γ elevation. There is macrophage accumulation and hyperactivity, along with B-cell stimulation and hypergammaglobulinemia. TNF-α and GM-CSF promote fusion of activated macrophages into the multinucleated cells seen within the granuloma .
Monocyte chemotactic factor (MCF), produced by activated T helper cells, attracts monocytes from the circulation into peripheral tissues. Compartmentalization of granuloma-forming T lymphocytes and monocytes within peripheral tissues leads to lymphopenia and decreased delayed-type hypersensitivity to common antigens (anergy), most pronounced during the initial stages of sarcoidosis. In addition, T regulatory cells may play a role in this anergic state. However, in some patients, there actually may be inadequate regulatory T-cell function such that production of TNF-α or IFN-γ is not suppressed .
Triggers
The identity of the antigen responsible for the cascade of events leading to granuloma formation in patients with sarcoidosis remains uncertain. Some investigators have proposed an autoimmune etiology, while others have searched for exposures to inorganic dusts and particles (e.g. zirconium, talc) or an infectious cause. The latter is suggested by several observations, including the identification of microbial components such as mycobacterial DNA sequences in different sarcoidal tissues , the transmissibility of sarcoidal lesions via the Kveim reagent, and the observation that sarcoidal granulomas have developed in patients receiving organ transplants from donors with sarcoidosis. In a meta-analysis, mycobacterial nucleic acids were identified in ~25% of sarcoidal tissues . Also, the catalase-peroxidase (mKatG) protein from Mycobacterium tuberculosis has been proposed as a potential triggering antigen for granulomatous inflammation.
However, arguments against a purely infectious etiology include the observation that sarcoidosis patients do not develop fulminant infectious symptoms when placed on immunosuppressive agents. In addition, mycobacteria have not been cultured from sarcoidal tissues or from the sarcoid-like lesions that develop during antiretroviral-induced immune reconstitution . Occupational associations have been noted in healthcare workers, navy personnel on aircraft carriers, and firemen , with a marked increase in sarcoidosis-like illnesses in the first responders to the 9/11 World Trade Center terrorist attacks. Additional proposed triggers include viruses , Propionibacterium acnes , and an abnormal accumulation of serum amyloid A (SAA).
Over the past two decades, an increasing number of medications have been reported to induce cutaneous sarcoidosis and sarcoid-like granulomatous eruptions, including IFN, TNF inhibitors, immune checkpoint inhibitors (e.g. ipilimumab, nivolumab), and targeted kinase inhibitors (e.g. vemurafenib). Patients with lymphoma may also develop secondary sarcoidal reactions, most commonly in lymph nodes but occasionally in the skin.
Genetics
Sarcoidosis is a polygenic not a monogenic disease. A genetic component is supported by a twin study in which the monozygotic twins of patients with sarcoidosis had an 80-fold increased risk of having sarcoidosis . The largest case–control study to date demonstrated a familial relative risk of 4.7 . The impact of HLA alleles varies significantly, depending upon disease subtype and racial group. Overall, HLA-DRB1*01 and DRB1*04 protect against sarcoidosis in some populations, while DRB1*03, DRB1*11, DRB1*12, DRB1*14, and DRB1*15 appear to confer risk ; HLA-B8/DR3 may be associated with the development of Löfgren syndrome (see below). In African-Americans, HLA-DQB1 alleles may play a role , and patients with DRB1*03 may have a better prognosis. Specific susceptibility genes include TNF (variants associated with Löfgren syndrome) and IL23R (variants confer risk for Crohn disease and sarcoidosis) .
While the etiologic agent clearly remains elusive, investigation of the immunopathogenesis of the disease (see above) has been aided by examining the cutaneous reaction produced by injection of a tissue suspension prepared from sarcoidal spleen. The suspension (Kveim–Siltzbach antigen) causes characteristic non-caseating granuloma formation in the skin of patients with sarcoidosis . Nowadays, however, this skin test is rarely performed.
Clinical Features
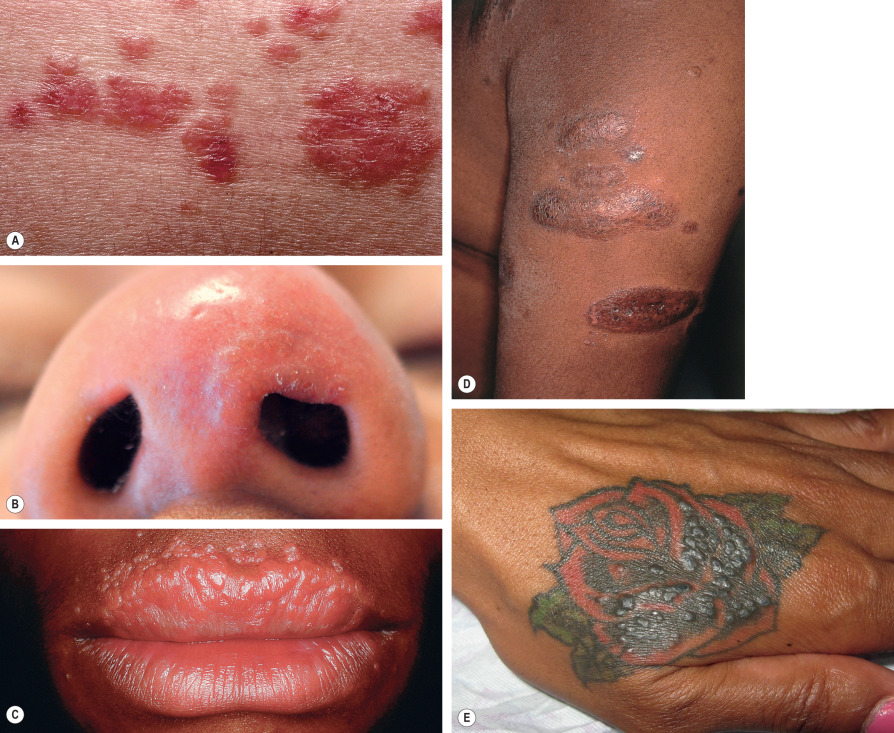
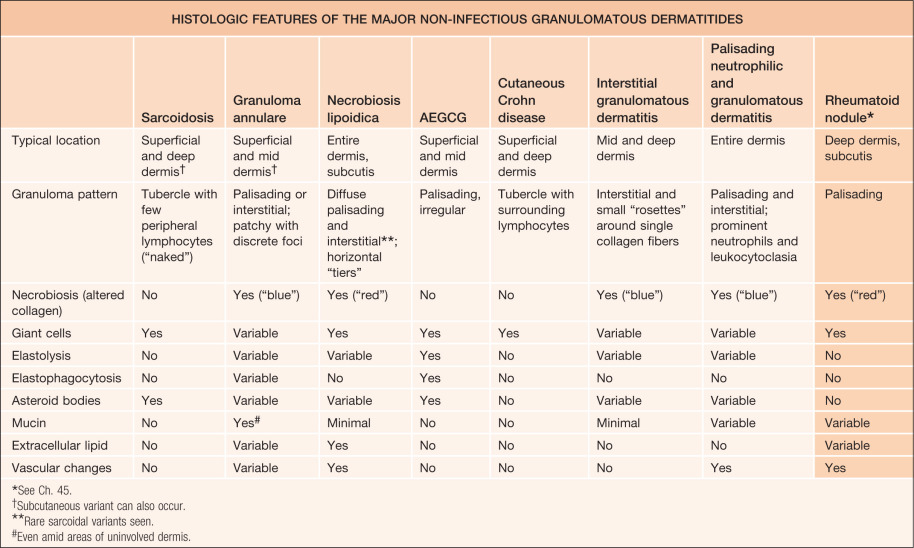
Up to a third of patients with systemic sarcoidosis develop skin lesions, which may be the first or only clinical manifestation of the disease. Although cutaneous sarcoidosis most commonly presents as papules and plaques, often red–brown to violaceous in color ( Figs 93.2A–D ), the skin manifestations are protean ( Table 93.2 ). Sarcoidal lesions are fairly symmetric in distribution and favor the nose, especially the alae, and the periocular and perioral regions of the face, followed by the neck, upper trunk, and extremities. Involvement at sites of prior injuries, e.g. scars, tattoos, is a characteristic finding ( Fig. 93.2E ). Unusual variants include hypopigmented, ichthyosiform, micropapular, psoriasiform, and ulcerative ( Fig. 93.3A,B ; see Table 93.2 ).
| SPECTRUM OF CUTANEOUS MANIFESTATIONS OF SARCOIDOSIS | |
|---|---|
| Common | |
|
|
|
|
| |
| Uncommon | |
|
|
|
|
| |
| Rare | |
|
|
|
|
|
|
|
|
|
|
| Nonspecific and common | |
| |
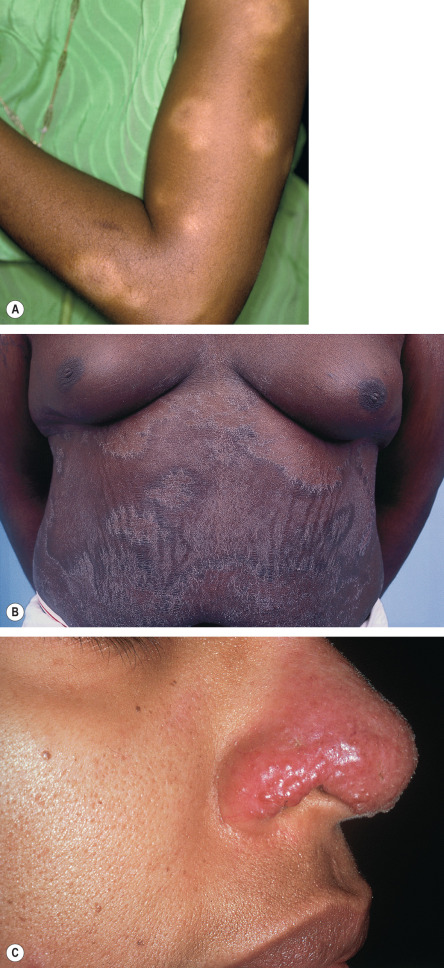
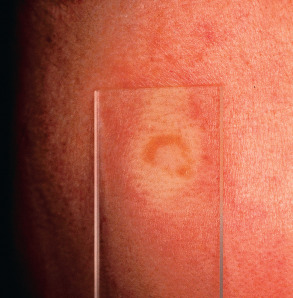
Although many lesions are red–brown to violaceous in color, they can vary from yellow–brown to erythematous, especially in lightly pigmented skin. Upon diascopy, in which pressure induces blanching, the lesions are said to have the color of “apple jelly” ( Fig. 93.4 ); this finding is usually easier to appreciate in lightly pigmented skin. Individual plaques can develop central clearing leading to an annular configuration or they can contain prominent telangiectasias (angiolupoid sarcoidosis). Dermoscopic features include small translucent yellow to orange globules, linear vessels, and central scar-like areas.
Additional variants of sarcoidosis of relevance to dermatologists include Löfgren syndrome, Darier–Roussy disease, and lupus pernio. Patients with Löfgren syndrome present acutely with fever, arthritis, hilar adenopathy, and erythema nodosum. They often require supportive measures and occasionally systemic corticosteroids, with spontaneous resolution occurring over 1–2 years. Patients with Darier–Roussy sarcoidosis present with painless, firm, subcutaneous nodules or plaques without epidermal changes. This variant represents sarcoidosis limited to the panniculus and is often associated with systemic sarcoidosis. Lupus pernio is characterized by papulonodules and plaques, which are often on the nose and cheeks and have a violaceous color, along with scale; there may be a beaded appearance along the nasal rim (see Fig. 93.3C ). Recognition of lupus pernio is important because of its association with chronic sarcoidosis of the lungs (~75% of patients) and of the upper respiratory tract (~50% of patients).
The most important nonspecific cutaneous manifestation of sarcoidosis is erythema nodosum ( Fig. 93.5 ). Erythema nodosum is associated with subacute, transient sarcoidosis that usually resolves spontaneously and typically does not require systemic corticosteroid therapy. In general, there are no additional cutaneous manifestations.
Nail changes can be seen in sarcoidosis, including clubbing, subungual hyperkeratosis, and onycholysis. Oral sarcoidosis may affect the soft mucosa, gingival tissue, tongue, hard palate, and major salivary glands. Heerfordt syndrome (uveoparotid fever) includes parotid gland enlargement, uveitis, fever, and cranial nerve palsies, usually of the facial nerve.
Systemic manifestations of sarcoidosis are also protean. Lung disease occurs in ~90% of patients, ranging from alveolitis to granulomatous infiltration of the alveoli, blood vessels, bronchioles, pleura, and fibrous septa . The end stage of pulmonary sarcoidosis is fibrosis with bronchiolectasis and “honeycombing” of the lung parenchyma. Hilar and/or paratracheal lymphadenopathy, which is usually asymptomatic, occurs in 90% of patients. Additional systemic manifestations are outlined in Table 93.3 .
| SYSTEMIC MANIFESTATIONS OF SARCOIDOSIS | |||
|---|---|---|---|
| Organ | % of patients affected | Clinical manifestations/radiographic and laboratory findings | Evaluation |
| Lungs | 90–95 | Dyspnea, non-productive cough/pulmonary infiltrates, fibrosis, restrictive lung disease (↓VC, ↓RV, ↓TLC, ↓DLCO) | CXR, high resolution chest CT scan (more sensitive than CXR), PFTs that include DLCO |
| Lymph nodes (LN) | 30–40 * | Lymphadenopathy/enlarged hilar and/or paratracheal LN | CXR, high resolution chest CT scan (more sensitive than CXR) |
| Eyes | 25 | Uveitis (can be asymptomatic despite being severe), conjunctivitis, sicca symptoms | Yearly ophthalmologic examination |
| Liver/spleen | 10–20 | Hepatomegaly and/or splenomegaly (rarely clinically relevant), cirrhosis, consequences of splenic enlargement (e.g. thrombocytopenia)/↑LFTs, ↓plts |
|
| Heart | 25 (5% clinically relevant) | Palpitations, sudden death, CHF/arrhythmias, cardiomegaly |
|
| CNS and peripheral nervous system | 10–20 | Neuropathies – cranial, spinal cord, peripheral, small fiber |
|
| Upper respiratory tract, including sinuses | 5–10 | Sinusitus, nasal congestion, stridor, parotiditis | Referral to otolaryngologist and/or dedicated imaging |
| Bones | 5–10 | Usually asymptomatic/lytic bone lesions | Radiography |
| Joints/muscles | 5–10 | Arthritis, weakness (up to a third of patients have severe fatigue), myopathy |
|
| Bone marrow | 50 |
| CBC, SPEP |
| Kidneys | 10–40 | Nephrolithiasis/hypercalciuria, ↓renal function |
|
| Endocrine | 5–10 |
|
|
| Other | <1 (rare) |
| Site-specific imaging |
* Pertains to peripheral lymphadenopathy; up to 90% have hilar lymphadenopathy.
** An association is based primarily on two studies, one of which found two cases of lymphoma in 2544 patients with sarcoidosis and the other had a standardized incidence ratio of 1.52 with a CI of 1.12–2.02. Multiple other studies have failed to find an association.
Childhood sarcoidosis is rare, and it usually presents with a triad of arthritis, uveitis and cutaneous lesions, along with constitutional symptoms. Peripheral lymphadenopathy is frequently present, but pulmonary involvement is less common than in adults. If sarcoidosis is being considered in a child, it is important to exclude Blau syndrome (see Ch. 45 ).
Pathology
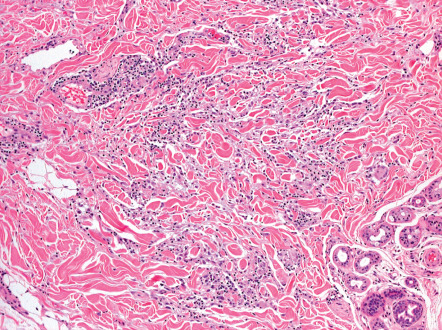
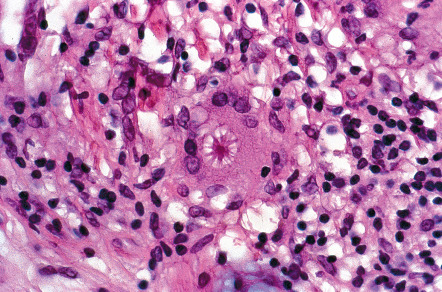
The histopathologic hallmark of sarcoidosis is the presence of superficial and deep dermal epithelioid cell granulomas devoid of prominent infiltrates of lymphocytes or plasma cells (“naked tubercles”) ( Fig. 93.6 ). Central caseation is usually absent, although fibrinoid deposition may be observed in up to 10% of cases. Multinucleated histiocytes (“giant cells”) are usually of the Langhans type, with nuclei arranged in a peripheral arc or circular fashion. The giant cells may contain eosinophilic stellate inclusions known as asteroid bodies ( Fig. 93.7 ) or rounded laminated basophilic inclusions known as Schaumann bodies, although neither is specific or required for the diagnosis. Asteroid bodies represent engulfed collagen, whereas Schaumann bodies likely represent degenerating lysosomes. Notably, up to 20% of biopsies of sarcoidosis contain polarizable material; therefore its presence does not exclude the diagnosis. In vulvar sarcoidosis, transepidermal elimination of the granulomas may be seen.
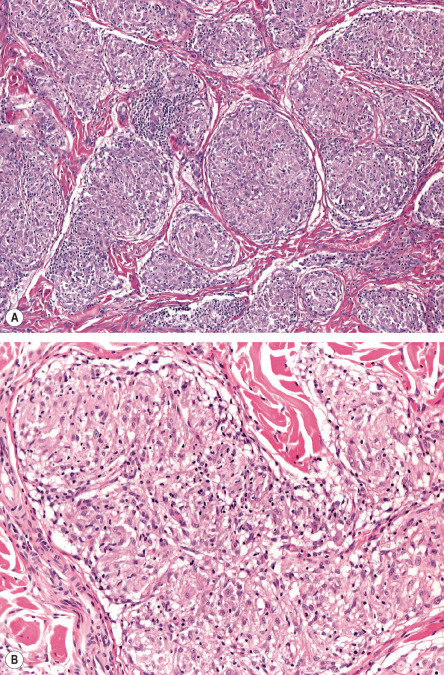
A range exists within the histologic spectrum of sarcoidosis, from the characteristic tubercles with minimal or no surrounding lymphocytic inflammation to unusual cases with dense lymphocytic and plasmacytic infiltrates around and within the nodular histiocytic aggregates. Occasionally, these aggregates may extend into the subcutaneous fat, producing the clinical features of Darier–Roussy sarcoidosis.
Diagnosis and Differential Diagnosis
Sarcoidosis is a diagnosis of exclusion, both clinically and histologically. In order to establish the diagnosis, a supportive clinical history must be accompanied by the histologic presence of non-caseating granulomas in at least one organ system. Details of systemic manifestations are outlined in Table 93.3 . An algorithm for initial and longitudinal evaluation has been proposed .
Serologically, elevated antinuclear antibody titers occur in ~30% of patients. The serum angiotensin-converting enzyme (ACE) level is elevated in ~60% of patients; it has a false-positive incidence of 10%, making it a more useful test for monitoring disease progression than for establishing the diagnosis. An ACE level >2–3 times the upper limit of normal is more suggestive of sarcoidosis.
Like syphilis, sarcoidosis is a great mimic and the clinical differential diagnosis depends upon the type of presenting clinical lesions. For example, a list of other disorders, in addition to sarcoidosis, that can present with annular lesions is provided in Table 19.1 . Papules, nodules and plaques may be nonspecific clinically, dictating a biopsy, and then the differential diagnosis rests upon the histopathologic findings. It is important to exclude the possibility of drug-induced cutaneous sarcoidosis (e.g. IFN-α for hepatitis C viral infection, TNF-α inhibitors).
The histologic differential diagnosis is broad, and includes multiple infections that lead to granulomatous inflammation. Special stains for acid-fast and fungal organisms should be obtained. When clinically appropriate, tissue culture should be performed. Increasingly, PCR is being utilized to exclude infections, including mycobacterial. Both tuberculoid leprosy and lupus vulgaris are in the differential diagnosis and for the latter, a QuantiFERON ® -TB Gold test may provide additional information. It is especially important to exclude infectious etiologies before initiating immunosuppressive agents.
Other histologic mimics include foreign body reactions to zirconium, beryllium, silica, tattoo ink, or soft tissue fillers. Special laboratory techniques, including histochemical, microincineration or spectrophotometric examinations, may be required to identify the causative agents (see Table 94.3 ). Specimens should be polarized to exclude birefringent foreign material as a causative agent, although up to 20% of known sarcoidal granulomas will have foreign material present within them. This finding is particularly common in lesions from the elbows and knees. Therefore, the dermatologic diagnoses of foreign body granuloma and sarcoidosis are not mutually exclusive .
In addition, granulomatous mycosis fungoides, Hodgkin disease, granulomatous rosacea, cutaneous Crohn disease, Blau syndrome (see Ch. 45 ), cheilitis granulomatosa, and the sarcoidal reaction to an underlying lymphoma can have histologic appearances similar to sarcoidosis. The histologic differences between sarcoidosis, granuloma annulare, annular elastolytic giant cell granuloma (AEGCG), necrobiosis lipoidica, rheumatoid nodule, and interstitial granulomatous dermatitis are outlined in Table 93.4 .
Treatment
Corticosteroids, in either topical, intralesional or systemic form, are the mainstay of therapy for systemic sarcoidosis ( Table 93.5 ). Therapy is guided by disease severity and progression. The typical oral prednisone dose for systemic disease is 0.5–1 mg/kg/day for 4–6 weeks, followed by a slow taper over months to years as dictated by the pulmonary disease, sarcoidosis of the upper respiratory tract, ocular disease, or other internal manifestations. The goal is alternate-day prednisone at the lowest possible dose that will maintain a remission. Cutaneous sarcoidosis may respond from the onset to lower doses of prednisone as well as every-other-day regimens. For limited cutaneous disease, topical or intralesional corticosteroids may suffice.
For treatment of more widespread cutaneous disease, alternative nonsteroidal systemic medications include hydroxychloroquine (200–400 mg/day), chloroquine (250–500 mg/day), minocycline (200 mg/day), methotrexate (10–25 mg weekly), and thalidomide (50–300 mg/day). Improvement of both systemic and cutaneous sarcoidosis has been observed with TNF-α inhibitors, including infliximab and adalimumab . These agents may be particularly effective for chronic, recalcitrant sarcoidosis (e.g. lupus pernio), but notably TNF-α inhibitors occasionally can trigger sarcoidosis . A treatment algorithm, based upon evidence-based methodology, has been proposed .
Granuloma Annulare
▪ Pseudorheumatoid nodule – subcutaneous granuloma annulare variant ▪ Generalized granuloma annulare – disseminated granuloma annulare
- ▪
Small grouped papules assuming an annular configuration, often in a symmetrical and acral distribution
- ▪
Seen primarily in children and young adults
- ▪
Clinical variants include localized, generalized, and subcutaneous forms
- ▪
Possible associations with systemic diseases, including diabetes mellitus, hyperlipidemia, thyroid disease, and rarely HIV infection, remain controversial
- ▪
Histopathologically, there is an infiltrative or palisading granulomatous dermatitis with focal degeneration of collagen and elastin and deposition of mucin
History
Calcott Fox first described “ringed eruption of the fingers” in 1895. Radcliffe-Crocker called this entity granuloma annulare in 1902.
Epidemiology
Two-thirds of patients with granuloma annulare (GA) are <30 years of age. The female-to-male ratio is approximately 2 : 1 in this common disease.
Pathogenesis
The etiology of GA is unknown. Trauma, insect bite reactions, tuberculin skin testing, vaccination, UV exposure, and Borrelia and viral infections have all been proposed as inciting factors . Based on the T-cell subpopulations identified in GA lesions, a delayed-type hypersensitivity reaction to an unknown antigen has been postulated as the precipitating event . Morphologic similarities to other granulomatous processes suggest that GA is caused by a Th1 inflammatory reaction, with elevations in IL-2R-positive lymphocytes, IFN-γ-producing lymphocytes, and TNF production by macrophages . These lymphocytes release cytokines, including macrophage inhibitory factor, that cause monocytes to accumulate within the dermis and release lysosomal enzymes that can degrade connective tissue . One ultrastructural study found that the main alteration in GA is elastic fiber degeneration and suggested that this disease is primarily a disorder of elastic tissue injury .
Familial cases of GA have been reported, including cases in identical twins, and one small study suggested that an association may exist between generalized GA and HLA-Bw35 . Vessel-based mixed inflammatory cell infiltrates with endothelial swelling are sometimes detected in routine histopathologic specimens, and direct immunofluorescence studies of GA may show vessel-based deposits of immunoreactants, but a role for these findings in the pathogenesis of this disease has not been established.
Clinical Features
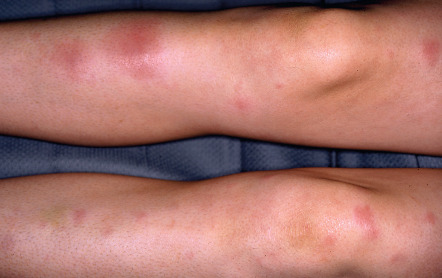
GA is a benign, usually self-limited, cutaneous disease that presents as arciform to annular plaques on the dorsal hands or feet of young people ( Fig. 93.8 ) . However, there is a wide array of clinical presentations, from a papular form to lesions that are subcutaneous, macular (patch form), or have central perforation ( Figs 93.9 & 93.10A ). Distribution patterns also vary, from localized to occasionally photodistributed or generalized/disseminated ( Fig. 93.10B ) . In general, the distribution of GA is: 60% isolated to the hands and arms, 20% on the legs and feet, 7% on both upper and lower extremities, 5% on the trunk, and 5% on the trunk plus other areas . Facial lesions are rare. The plaques may be skin-colored, pink or violaceous in color, and, upon close inspection, are found to be composed of individual small papules measuring a few millimeters in diameter. The lesions are usually asymptomatic.
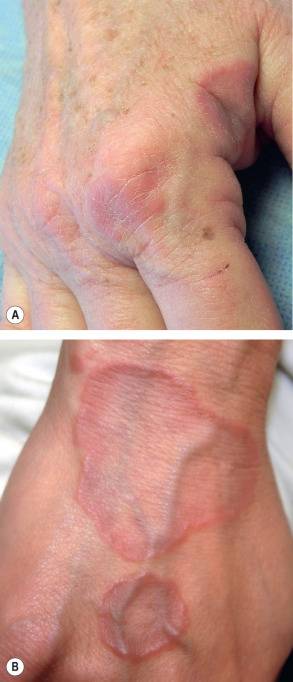
In generalized/disseminated GA, the widespread, symmetrically distributed lesions can be annular plaques or discrete papules and they favor the trunk and/or extremities. This form is uncommon and has a later age of onset, poorer response to therapy, and an increased prevalence of the HLA-Bw35 allele. In one study of 100 patients with generalized GA, 45% had lipid abnormalities, including hypercholesterolemia, hypertriglyceridemia or both, while ~20% had diabetes mellitus .
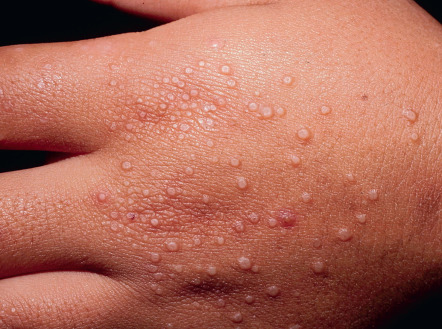
Deep dermal or subcutaneous GA manifests as large, painless, skin-colored nodules which may be mistaken for rheumatoid nodules, leading to the term pseudorheumatoid nodule. It has a predilection for children <6 years of age. Typical locations include the palms, hands, anterior tibial surfaces and feet, as well as the buttocks, scalp and, rarely, the eyelids . As many as 50% of individuals with deep GA lesions also have associated classic lesions.
In perforating GA, the small papules have central umbilication, scale-crust or focal ulceration, and they occur primarily on the dorsal hands and fingers (see Fig. 93.10A ). This variant occurs up to 5% of patients with GA, and histologically transepidermal elimination of degenerating collagen is seen . While umbilicated GA has been described, it probably represents a variant of perforating GA.
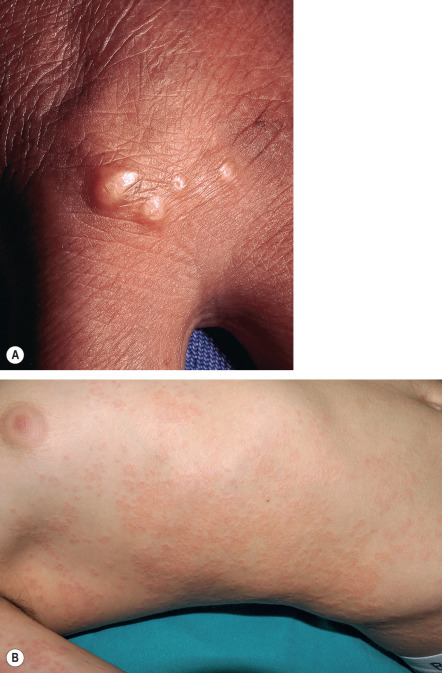
Patch GA is a distinct, unusual variant that is characterized by patches of erythema on the extremities and trunk. Symmetrical lesions on the dorsum of the feet often present as macular (i.e. “patch”) disease. Although an annular configuration may be absent, presence of the classic histopathologic findings of interstitial GA (see below) allow for the diagnosis to be made microscopically.
GA has been described as a paraneoplastic granulomatous reaction to solid organ tumors, Hodgkin disease, non-Hodgkin lymphoma, and granulomatous mycosis fungoides . In these patients, the clinical pattern is frequently atypical, with painful lesions in unusual locations, including the palms and soles. In a recent review of the literature, a relationship between GA and malignancy was not identified; however, patients with atypical presentations may warrant a complete physical exam with focused, age-appropriate screening . While photodistributed GA has been reported, some patients may have had AEGCG, which some clinicians view as a variant of GA.
Many reports either supporting or refuting the association of GA with diabetes mellitus have been published, but to date there have been no large-scale epidemiologic studies. In a retrospective study of 84 patients, 12% were found to have diabetes mellitus, and these patients were more likely to suffer from chronic relapsing GA than were non-diabetic patients . In a larger retrospective study of 1383 patients, diabetes mellitus was diagnosed in ~20% of patients with generalized GA, compared with ~10% of patients with localized GA . However, in a case–control study of 126 patients, an association was not identified . There have also been a few reports of an association with thyroid disease.
Classic GA and perforating GA may develop within herpes zoster scars (see Table 80.7 ) and following BCG vaccinations. Atypical variants of GA, including photodistributed, have been associated with HIV infection . Additional reported associations, based upon small studies, include hepatitis B virus and hepatitis C virus . Lastly, the possibility of drug-induced GA, most often due to TNF-α inhibitors, should be excluded; less common culprits include amlodipine, allopurinol, diclofenac, and gold .
Pathology
GA is a granulomatous dermatitis characterized by focal degeneration of collagen and elastic fibers, mucin deposition, and a perivascular and interstitial lymphohistiocytic infiltrate in the upper and mid dermis. The key to the histopathologic diagnosis of GA is the presence of mucin plus the identification of histiocytes in one of two patterns. The most common (accounting for ~50–70% of cases) is the interstitial or infiltrative pattern ( Fig. 93.11 ), in which scattered histiocytes are distributed between collagen fibers. Degeneration of collagen fibers is minimal, but granular, basophilic mucin deposition between collagen bundles can be highlighted with Alcian blue and colloidal iron stains . The second pattern (25–50% of cases) is more obvious and is easier to diagnose. It consists of one to several palisading granulomas with central connective tissue degeneration surrounded by histiocytes and lymphocytes ( Fig. 93.12 ). Mucin is abundant in the center of the palisaded granuloma, and fibrin, neutrophils and nuclear dust may also be present.