Abstract
Neutrophils are essential inflammatory cells. In response to infection they accumulate rapidly at sites of infection or injury and become activated. These cells are packed with granules whose contents kill and degrade target microorganisms following phagocytosis of opsonized microbes.
Occasionally, neutrophils can suddenly accumulate and become activated in the absence of an identifiable local infection. Release of their cytoplasmic granules then leads to injury of normal tissue. Neutrophilic dermatoses, including those seen in the setting of autoinflammatory disorders, are examples of this phenomenon. Examples of neutrophilic dermatoses include Sweet syndrome, pyoderma gangrenosum (PG), and Behçet disease. Both Sweet syndrome and PG can be idiopathic in nature but a significant proportion of patients have associated underlying diseases, e.g. hematologic malignancies, inflammatory bowel disease. Occasionally, these neutrophilic disorders are triggered by medications.
In addition to Sweet syndrome, PG and Behçet disease, this chapter reviews the clinical manifestations of bowel-associated dermatosis–arthritis syndrome and SAPHO ( s ynovitis, a cne, p ustulosis, h yperostosis and o steitis) syndrome.
Keywords
neutrophilic dermatoses, Sweet syndrome, acute neutrophilic dermatosis, pyoderma gangrenosum, Behçet disease, bowel-associated dermatosis–arthritis syndrome, autoinflammatory disorders, SAPHO ( s ynovitis, a cne, p ustulosis, h yperostosis and o steitis) syndrome
The neutrophilic dermatoses constitute a heterogeneous but linked spectrum of diseases , with significant overlapping histopathologic findings and similar pathogenic mechanisms and therapeutic approaches. They are often associated with underlying internal diseases, which may have significant morbidity and mortality. Histologically, these disorders are characterized by perivascular and diffuse neutrophilic infiltrates without any identifiable infectious agents ( Fig. 26.1 ). The cutaneous manifestations vary, from urticarial plaques and vesiculopustules to nodules and ulcers. It is noteworthy that there may be several different types of lesions in the same patient. The dermatosis may be localized or more widespread, and, in an occasional patient, similar sterile neutrophilic infiltrates may occur in other organs including the eyes, joints, bones, lung and kidney. The neutrophilic infiltrate may be most prominent in the epidermis, dermis, or even the panniculus. Distinct entities have been defined by the nature of their clinical and histologic presentations as well as their associated diseases.
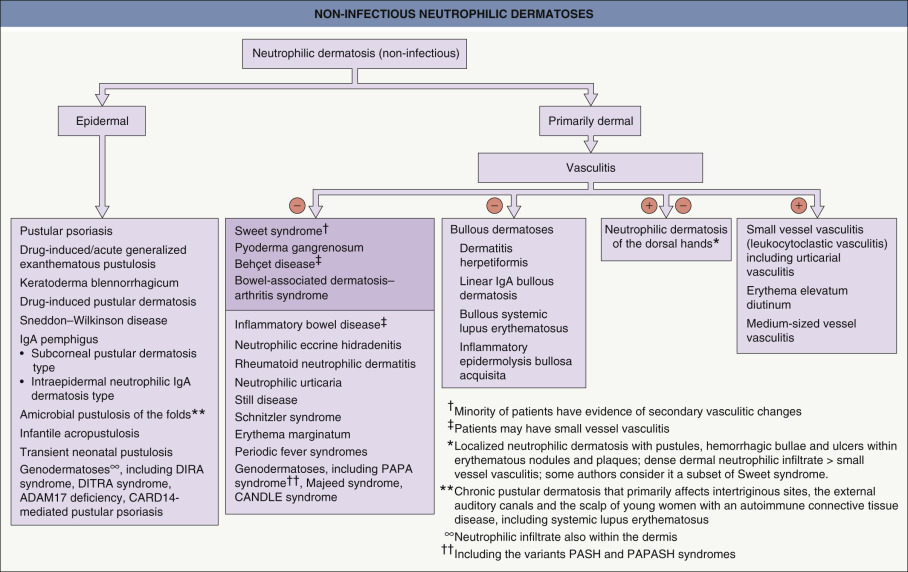
Of note, patients with autoinflammatory disorders may develop neutrophilic dermatoses, from sterile pustules in DIRA (deficiency of IL-1 receptor antagonist) to pyoderma gangrenosum in PAPA (pyogenic [sterile] arthritis, pyoderma gangrenosum, and acne) and PASH (pyoderma gangrenosum, acne, and suppurative hidradenitis) syndromes (see Tables 45.6 and 45.7 ). There is also overlap between neutrophilic dermatoses and neutrophilic urticaria (see Table 45.6 ).
Neutrophil Biology
Granulocytes (neutrophils, eosinophils and basophils) are cells essential to our defense against microbes as well as other inflammatory responses. The neutrophil is a terminally differentiated, non-dividing cell which is packed with granules whose contents kill and degrade target microorganisms. Recent advances in our understanding of neutrophil biology include the molecular and cellular mechanisms responsible for their production and release from the bone marrow (e.g. chemokine CXC receptor 4 [CXCR-4]; see Ch. 60 ); their recruitment, priming and activation within inflamed tissues ( Table 26.1 ); and the events resulting in their removal.
| NEUTROPHILS – THEIR RELATIONSHIP TO SITES OF INFLAMMATION |
| Intravascular adhesion, activation and migration |
|
| Migration within tissue |
|
| Activation within tissue |
|
| Phagocytosis |
|
| Degranulation |
|
Granulocytes, including neutrophils, originate in the bone marrow from pluripotent cells. In order to supply sufficient numbers of circulating cells, neutrophils are produced within the bone marrow at a prodigious baseline rate (>5–10 × 10 10 neutrophils daily). The bone marrow also has the capacity to upregulate granulocyte production sharply in response to a number of stresses such as infection. Mature neutrophils circulate in the peripheral bloodstream for only 6–8 hours before migrating into tissues, where they survive for 2–3 days. Neutrophils, unlike platelets and erythrocytes, appear to be removed from the circulation in a random rather than an age-related manner.
Transcriptional profiling studies suggest that granulocytes arise via the selective expression of a subset of transcription factors (e.g. STAT3), granulocyte proteins (e.g. neutrophil elastase), and receptors (e.g. N-formyl-methionyl-leucyl-phenylalanine [fMLP]). Differentiation from pluripotent stem cells requires 7–10 days, and during this period, under the influence of cytokines, neutrophils acquire their characteristic appearance and granules (primary, secondary and tertiary). The following stages of myeloid maturation are recognized: myeloblast, promyelocyte, myelocyte, metamyelocyte, band, and, finally, the segmented neutrophil. The progressive gain of differentiated characteristics is accompanied by a loss in the potential to proliferate, i.e. beyond the myelocyte stage, the cells are non-dividing. The intracellular granules acquired during maturation contain enzymes that mediate the oxidative and non-oxidative killing functions of the neutrophil :
- •
primary (azurophilic) granules – acquired at the promyelocyte stage and their contents include myeloperoxidase, lysozyme, neutrophil elastase, defensins, proteinase 3, and bactericidal/permeability-increasing protein
- •
secondary granules – acquired at the transition to the myelocyte stage and their contents include lactoferrin, neutrophil collagenase, neutrophil gelatinase-associated lipocalin, and lysozyme
- •
tertiary granules – acquired during later stages of neutrophil maturation and contain neutrophil gelatinase and leukolysin.
Inflammation
A critical role of inflammation is to deliver neutrophils and other leukocytes to a site of injury and then activate these cells to perform their function of protecting the host against infection. Neutrophils are among the first cells to arrive at sites of inflammation. Reasons for this include their abundance in the bloodstream and their rapid response to chemokines. When activated, neutrophils move at speeds up to 30 microns/min – the fastest cell in the body.
The motile responses of neutrophils to microbial infection include emigration out of the vasculature and movement toward the source of the inflammatory chemoattractant. This culminates in the phagocytic ingestion of opsonized microbes. In order to arrive at the site of infection, leukocytes must migrate out of the vasculature via margination, rolling, activation, and tight adhesion (see Ch. 102 ). They then move toward the site of injury or infection (see Table 26.1 ) and eventually undergo degranulation then apoptosis.
The price to be paid for the defensive potency of neutrophils for destroying microbes and necrotic tissues is that they can injure normal tissue. During activation and phagocytosis, neutrophils release products (e.g. lysosomal enzymes, reactive oxygen intermediates, products of arachidonic acid metabolism [prostaglandins and leukotrienes]) not just within the phagolysosome, but also into the extracellular space. Endothelial injury and tissue damage ensue, thus contributing to a number of acute and chronic diseases that affect the skin as well as other organs.
Sweet Syndrome
▪ Acute febrile neutrophilic dermatosis
- ▪
Constitutional signs and symptoms such as fever and malaise
- ▪
Clinically, erythematous plaques that are occasionally bullous
- ▪
Histologically, dense perivascular neutrophilic infiltrate, edema and, infrequently, bullae; leukocytoclasia with minimal to no evidence of vasculitis
- ▪
Associated conditions include infections, malignancies (especially acute myelogenous leukemia), inflammatory bowel disease, autoimmune disorders (e.g. systemic lupus erythematosus [SLE]), drugs, and pregnancy
History
The prototype of the neutrophilic dermatoses is Sweet syndrome. In 1964, Sweet described eight middle-aged women who had an acute onset of fever and erythematous plaques associated with a nonspecific infection of the respiratory tract or gastrointestinal tract. Histologically, the skin lesions were characterized by a neutrophilic infiltrate. He named this constellation of findings “acute febrile neutrophilic dermatosis”. In 1968, Whittle and colleagues reported a similar case and named it “Sweet’s syndrome”.
Epidemiology
Sweet syndrome is an uncommon disease, with a worldwide distribution and no obvious racial predilection, although the disorder appears to be more frequent in Japan. The average age of onset is 30–60 years, but infants, children and the elderly may be affected; there is a female predominance of 4 to 1. At least half of patients have an identifiable associated disorder or trigger: 15–30% have an internal malignancy (hematologic ≫ solid organ), ~25% a preceding infection, and ~10% exposure to a potentially causative drug ( Table 26.2 ) . While the epidemiologic statistics in part reflect associated underlying disorders including internal malignancies, inflammatory bowel disease and autoimmune connective tissue diseases such as SLE , they do not completely explain the female predominance.
| SWEET SYNDROME – ASSOCIATED DISORDERS AND TRIGGERS |
| Infections |
|
| Malignancies |
|
| Gastrointestinal disorders |
|
| Drugs |
|
| Autoimmune disorders |
|
* Includes acute and chronic myelogenous leukemia, chronic lymphocytic leukemia, non-Hodgkin disease, Hodgkin’s disease, myeloma, other myeloproliferative disorders.
‡ Some authors use the terms non-bullous neutrophilic LE or neutrophilic dermatosis in conjunction with LE rather than Sweet syndrome associated with SLE.
Pathogenesis
The pathogenesis of Sweet syndrome is unknown. The association with underlying diseases suggests a hypersensitivity reaction. One hypothesis is a local or systemic dysregulation of cytokine secretion, involving interleukin (IL)-1, granulocyte colony-stimulating factor (G-CSF), granulocyte–macrophage colony-stimulating factor (GM-CSF), and interferon-γ . For example, increased expression of IL-1β has been noted within cutaneous lesions. Defects in transcriptional regulation of hematopoietic PTPN6 , which encodes protein tyrosine phosphatase nonreceptor type 6, and formation of splice variants are also thought to be involved in the pathogenesis of Sweet syndrome . Lastly, in some patients, mutations have detected in the gene associated with familial Mediterranean fever, MEFV .
Clinical Features
The initial cutaneous lesions are tender, non-pruritic, erythematous plaques or papules, which may enlarge or coalesce to form plaques with an uneven mammillated surface ( Figs 26.2 & 26.3 ). Because of the pronounced associated edema, the lesions may have a pseudovesicular or pseudopustular appearance; however, some patients actually do develop vesiculation, bullae or pustules within their plaques. Occasionally, the plaques have a central yellowish discoloration creating a targetoid appearance. A facial erysipelas-like appearance has also been described as has a giant cellulitis-like variant. The vesiculobullous variant, which is most frequently associated with acute myelogenous leukemia, can progress to ulceration resembling superficial pyoderma gangrenosum (PG). In this form, there may be a single lesion or multiple, asymmetrically distributed lesions.
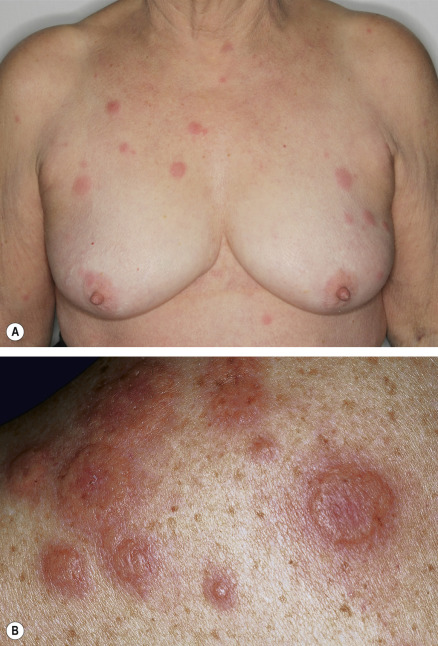
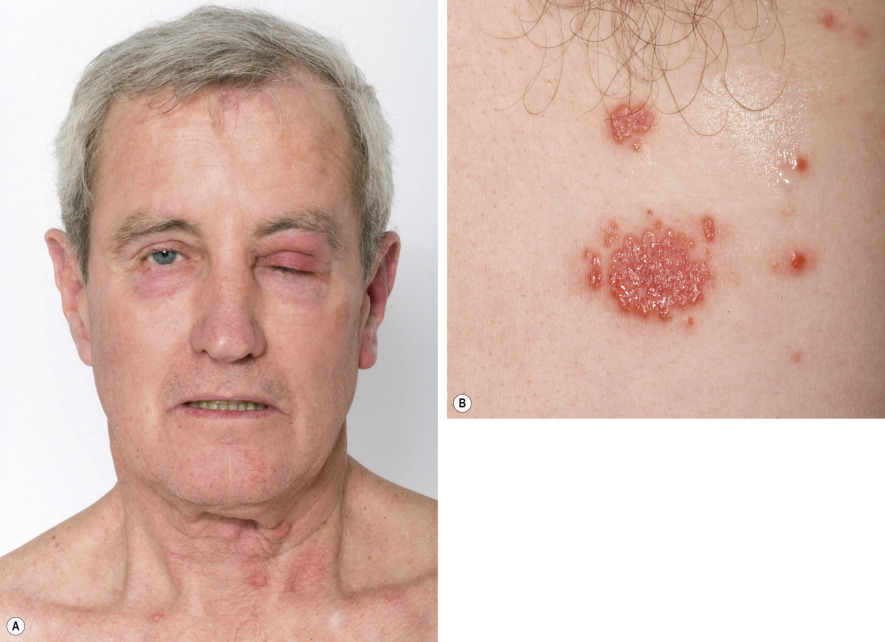
The cutaneous eruption of Sweet syndrome favors the head, neck and upper extremities (including the dorsal aspect of the hands), but can occur anywhere. In true malignancy-associated cases, the lesions tend to have a more widespread distribution. Papulonodules involving the lower legs may resemble erythema nodosum and although erythema nodosum has been reported in patients with Sweet syndrome, such nodules more likely represent a neutrophilic panniculitis. When isolated, the latter is often referred to as subcutaneous Sweet syndrome.
As with PG, specific lesions may be initiated by wounding injuries such as needle sticks (pathergy). Oral lesions are uncommon, except in patients with hematologic disorders; initially they appear pseudopustular, and later ulcerate and appear as aphthae. The cutaneous eruption of Sweet syndrome usually resolves spontaneously within 5–12 weeks but recurs in up to 30% of patients.
An upper respiratory tract infection or flu-like illness frequently precedes the development of the syndrome. Fever occurs in 40–80% of patients and can be intermittent. Extracutaneous involvement is also frequently seen ( Fig. 26.4 ; Table 26.3 ).
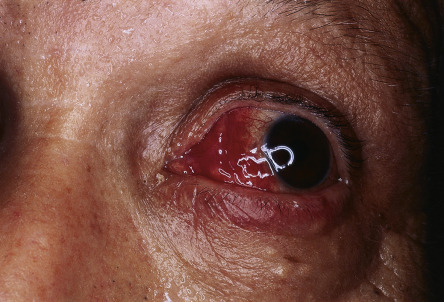
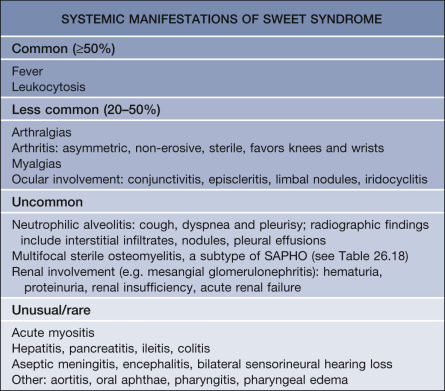
Associated Diseases
These are summarized in Table 26.2 and should guide the history, including all medications, physical examination, and focused laboratory evaluation .
Pathology
The histopathologic features are contingent upon the type of cutaneous lesion sampled. The characteristic histologic presentation is a diffuse nodular and perivascular neutrophilic infiltrate without evidence of vasculitis ( Fig. 26.5 ), although occasionally leukocytoclastic vasculitis (LCV) can be observed. Leukocytoclasia with endothelial swelling, but without the fibrinoid necrosis that fulfills the criteria for LCV, is the usual finding. Occasionally, the dermal infiltrate may extend into the subcutis, creating a septal or, less frequently, a lobular panniculitis. Isolated neutrophilic panniculitis has been described.
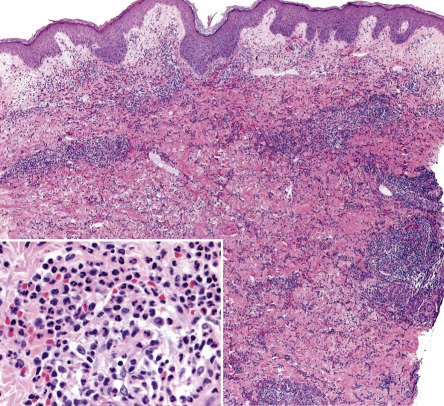
In general, epidermal changes are not significant. However, neutrophils occasionally invade the epidermis, producing subcorneal pustules, and they may also infiltrate the adnexa. In the setting of significant edema, there can be epidermal spongiosis and sometimes reticular degeneration as well as intraepidermal and subepidermal vesiculation.
Three histologic variants of Sweet syndrome, histiocytoid, lymphocytic and eosinophilic, have been increasingly recognized. The histiocytoid variant is characterized by a dermal, and sometimes subcutaneous, infiltrate composed of histiocyte-like immature myeloid cells . These cells have myeloperoxidase activity ( Fig. 26.6 ) and must be distinguished from those of leukemia cutis. The lymphocytic variant is often associated with, and sometimes precedes, underlying myelodysplasia . More recently, an eosinophilic variant has been described .
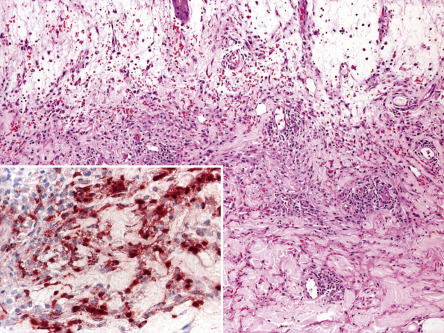
Differential Diagnosis
Although there are no specific diagnostic findings in this disease, a peripheral leukocytosis with neutrophilia and an elevated ESR and C-reactive protein are frequently seen. In patients with an associated hematologic malignancy or myelodysplasia, there may be a high or low white cell count, lymphocytosis or lymphopenia, and thrombocytosis or thrombocytopenia. Although elevated serum levels of antineutrophil cytoplasmic antibody (ANCA) have been reported , in particular pANCA or an atypical ANCA, there is no strong evidence that it is a serologic marker for this disease.
In 1986, Su and Liu proposed two major and four minor criteria for the diagnosis of Sweet syndrome, which have been widely accepted and are shown with minor revisions in Table 26.4 . The differential diagnosis of the syndrome depends on the nature and age of the lesions and on the associated disorder ( Table 26.5 ; see Fig. 26.1 ).
| CRITERIA FOR DIAGNOSIS OF SWEET SYNDROME |
| Major criteria |
|
| Minor criteria |
|
| THE DIFFERENTIAL DIAGNOSIS OF SWEET SYNDROME |
| Inflammatory dermatoses |
|
| Infections |
|
| Neoplasms |
|
Patients with neutrophilic dermatosis of the dorsal hands develop tender, erythematous to violaceous plaques that may become bullous or ulcerative ( Fig. 26.7 ) . There is a spectrum of associated histologic features, from pustular vasculitis to those of PG or Sweet syndrome. Hence, some authors consider this to be a variant of Sweet syndrome.
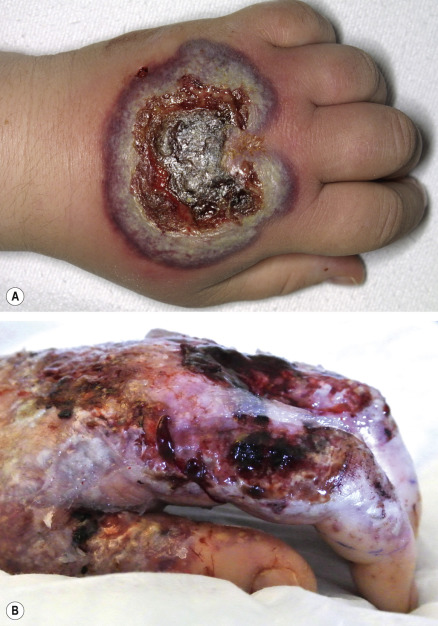
In patients with hematologic malignancies, especially acute myelogenous leukemia, several neutrophilic dermatoses, e.g. Sweet syndrome, atypical (bullous) pyoderma gangrenosum, and neutrophilic eccrine hidradenitis, can occur alone or in combination. The approach to such a patient is presented in Table 26.6 .
| NEUTROPHILIC DERMATOSES ASSOCIATED WITH HEMATOLOGIC MALIGNANCIES | |||
|---|---|---|---|
| Sweet syndrome | Atypical (bullous) pyoderma gangrenosum | Neutrophilic eccrine hidradenitis | |
| |||
| Painful plaques and nodules with occasional blisters and pustules within lesions | Hemorrhagic bullae and superficial ulcerations | Erythematous edematous plaques and papules which may be painful, infrequent pustules |
| Favors face, upper extremities and trunk | Favors face and upper extremities (especially dorsal hands) | Favors face, extremities, trunk | |
| Can recur (30–50% of patients) | Can recur | Resolves spontaneously over ~2 weeks without scarring Can recur | |
| Fever, arthralgia or arthritis Multiorgan involvement ( Table 26.3 ) Neutrophilia Elevated ESR | Systemic features usually absent | Fever, neutropenia * |
| Papillary edema with dermal neutrophilic infiltrate Histiocytoid variant – histiocyte-like immature myeloid cells Lymphocytic variant – in the setting of myelodysplasia | Subepidermal hemorrhagic bullae Pustules with neutrophilic dermal infiltrate | Neutrophilic † infiltrate about and within eccrine apparatus, necrosis of eccrine epithelium, and eccrine squamous syringometaplasia |
| Infections, hematologic malignancies > solid tumors, IBD, autoimmune diseases, pregnancy, G-CSF, all- trans -retinoic acid (see Table 26.2 ) | Hematologic disorders, especially acute myelogenous leukemia and myelodysplasia, IBD, G-CSF | Leukemia with or without chemotherapy; post-chemotherapy for lymphoma and solid tumors; G-CSF; rarely infections |
| Systemic corticosteroids (0.5–1 mg/kg/day for 4–6 weeks), potassium iodide (900 mg/day), dapsone (100–200 mg/day), colchicine (1.5 mg/day) (see Table 26.11 ) | Systemic corticosteroids (see Table 26.11 ) | None (spontaneous resolution) |
* Usually in setting of chemotherapy.
† May be lymphocytic in setting of chemotherapy-induced neutropenia.
In patients with rheumatoid arthritis, the possibility of rheumatoid neutrophilic dermatitis, including the uncommon annular variant, needs to be considered . As noted in Table 26.5 , Sweet syndrome can also mimic soft tissue infections, including cellulitis and necrotizing fasciitis . This distinction can prove challenging, especially in immunocompromised hosts.
Treatment
When limited to the skin, Sweet syndrome is a benign condition which, if left untreated, may persist for weeks or months. Cutaneous lesions then involute spontaneously, rarely leaving scars. However, recurrences develop in ~30% of patients (with or without treatment) and occur even more often in those with hematologic disorders (~50%). Although the initial clinical presentation often suggests sepsis, antibiotics, in general, are ineffective. However, when the disease is associated with a recognized infection, treatment of the underlying infection may result in improvement.
The most effective therapy for Sweet syndrome is oral prednisone (0.5–1.0 mg/kg/day) for 2–6 weeks. There is prompt relief of not only the cutaneous, but also the extracutaneous, manifestations. In some patients, prolonged low-dose prednisone for an additional 2–3 months may be necessary to suppress recurrences. When the lesions are few and localized, topical superpotent or intralesional corticosteroids may prove helpful.
The major alternative drugs are potassium iodide (900 mg/day; see Table 100.6 ), dapsone (100–200 mg/day), and colchicine (1.5 mg/day). They can also be used as corticosteroid-sparing agents. Nonsteroidal anti-inflammatory drugs (e.g. indomethacin, naproxen, sulindac), cyclosporine, thalidomide, lenalidomide, anakinra, methotrexate and rituximab have also been reported to lead to improvement of Sweet syndrome. Evidence for any of these therapeutic regimens is based on case reports or small case series. Of note, while TNF inhibitors have been used to treat refractory Sweet syndrome, they can also be a cause of drug-induced Sweet syndrome.
Pyoderma Gangrenosum
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


