Abstract
Neurofibromatosis type 1 (NF1) and tuberous sclerosis complex (TSC) are autosomal dominant neurocutaneous disorders characterized by hamartomas and tumors in the skin, eye, central nervous system, and other organs. Cutaneous lesions are often the initial clinical features in both diseases, and dermatologic evaluation frequently helps to establish the diagnosis. Recognition of the underlying genetic defects and the signaling pathways affected in these disorders has led to advances in molecular diagnosis and new targeted medical therapies. Approaches to distinguish NF1 and TSC from related conditions as well as current strategies for evaluation and management of affected individuals are highlighted.
Keywords
neurofibromatosis type 1, segmental neurofibromatosis, mosaic neurofibromatosis, café-au-lait macule, neurofibroma, plexiform neurofibroma, Lisch nodules, tuberous sclerosis complex, ash leaf macule, angiofibromas, ungual fibromas, shagreen patch
Introduction
Neurofibromatosis (NF) and tuberous sclerosis (TS) are neurocutaneous disorders, or phakomatoses. They are characterized by cutaneous lesions as well as peripheral and/or central nervous system neoplasms. Like most hereditary cancer syndromes, NF and TS are autosomal dominant in inheritance. Despite a high penetrance for both diseases, new cases of NF type 1 (NF1) and TS frequently represent de novo mutations. The pleiotropic nature of these two neurocutaneous syndromes has led to complex schemes for their classification and diagnosis. Dermatologists are frequently asked to participate in the clinical diagnosis of patients with NF1 and TS due to the early appearance of cutaneous, especially pigmentary, findings. Advances in the molecular characterization of these disorders have revealed related, but distinct, pathogenic mechanisms.
In 1982, Riccardi proposed a classification scheme for patients with NF ( Table 61.1 ). Elucidation of the molecular bases of NF1, NF2, segmental/mosaic NF1 (formerly known as NF5), and “NF1-like” syndromes has further refined the categorization of patients. Among these conditions, NF1 has the most well-characterized and diagnostic skin findings and is the main focus in this chapter.
| RICCARDI’S CLASSIFICATION OF NEUROFIBROMATOSIS | |
|---|---|
| Category | Description |
| NF1 | von Recklinghausen disease |
| NF2 | Acoustic schwannoma |
| NF3 * | Mixed (central and peripheral neurofibromas) |
| NF4 * | Variant |
| NF5 * , † | Segmental café-au-lait macules, neurofibromas, or both, affecting one or more dermatomes |
| NF6 * , ‡ | Café-au-lait macules only |
| NF7 * | Late onset |
| NF-NOS | Not otherwise specified |
* Primarily of historical/conceptual interest.
† Now referred to as mosaic (segmental) NF1.
Neurofibromatosis Type 1
▪ von Recklinghausen disease
- ▪
NF1 is an autosomal dominant disorder with an incidence of approximately 1 in 3000 births
- ▪
Characteristic clinical findings include café-au-lait macules, neurofibromas, freckling in the axillae and groin, Lisch nodules, and bony defects
- ▪
Patients with NF1 have an increased risk of developing juvenile myelomonocytic leukemia and a variety of extracutaneous tumors, including optic gliomas, malignant peripheral nerve sheath tumors, pheochromocytomas, and CNS tumors
- ▪
Up to 50% of patients with NF1 have a new spontaneous mutation rather than an inherited mutation
- ▪
The protein product of NF1 (neurofibromin) is involved in the negative regulation of Ras signaling
- ▪
Other well-characterized forms of NF include segmental/mosaic NF1 and NF2 (vestibular schwannomas)
History
Individuals with NF1 have been described throughout history . In 1882, von Recklinghausen provided the first in-depth report of the clinical and pathologic nature of the neurofibromas. Fifty years later, the Viennese ophthalmologist Lisch described the iris nodules that were more recently recognized as an important criterion in the clinical diagnosis of NF. In the mid twentieth century, Crowe, Schull, and Neel compiled a landmark monograph with a comprehensive description of the manifestations of this disease .
Epidemiology
NF1 occurs worldwide. Its incidence is approximately 1 in 3000 births .
Pathogenesis
NF1 is inherited in an autosomal dominant manner. Most large series have found that 30–50% of patients have no affected relatives and thus likely harbor new spontaneous mutations . There is also the possibility of germline mosaicism in a parent, especially when two siblings develop NF1 in the absence of a prior family history. The penetrance of NF1 approaches 100% in carefully examined adults , but expressivity is variable. A wide range of clinical findings and complications can develop among individuals with the same underlying mutation. The specific NF1 mutation is therefore not the only factor that determines phenotype.
In 1987, an international consortium found evidence of linkage to the pericentromeric region of chromosome 17 in families with NF1 . Subsequent descriptions of two NF1 patients with balanced translocations involving the long arm of chromosome 17 (17q11.2) further refined the position of the NF1 locus . The localization data derived from these studies led to the cloning of the NF1 gene .
The NF1 gene encodes neurofibromin, a 327 kDa protein composed of 2818 amino acids, and consists of at least 59 exons ( Fig. 61.1 ) . Over 80% of NF1 patients have a mutation that predicts truncation of the neurofibromin protein, and ~5% of patients have a large deletion (“microdeletion”) that encompasses the entire NF1 gene plus variable flanking genes . Although there is usually no clear genotype–phenotype correlation, exceptions include: (1) the single amino acid deletion p.Met922del and a missense mutation affecting p.Arg1809, each associated with an absence of neurofibromas and the latter with a Noonan syndrome-like phenotype ; and (2) a recurrent 1.4-mega-base pair deletion (“type 1 microdeletion”) resulting in a more severe phenotype . The latter includes an earlier onset and larger number of neurofibromas, an increased risk of malignant peripheral nerve sheath tumors, cognitive deficiencies, facial dysmorphism, overgrowth, and connective tissue dysplasia (e.g. hyperextensible skin, joint laxity) .
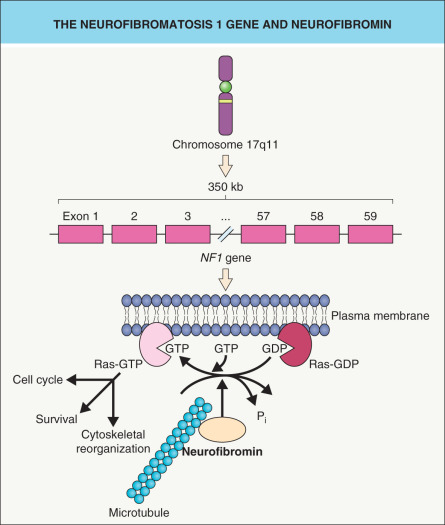
Sequence similarities between a central 300–400 amino acid domain within neurofibromin and G TPase- a ctivating p roteins (GAPs) led to recognition of neurofibromin’s role in the negative regulation of the Ras– m itogen- a ctivated p rotein k inase (MAPK) pathway that promotes cell survival and proliferation (see Fig. 61.1 and Fig. 55.4 ) . Like all G proteins, Ras activity requires GTP. Neurofibromin and other GAPs accelerate the hydrolysis of GTP to GDP, thereby terminating the Ras signal and functioning as tumor suppressor proteins. Neurofibromin also associates with microtubules , although the consequences remain unclear.
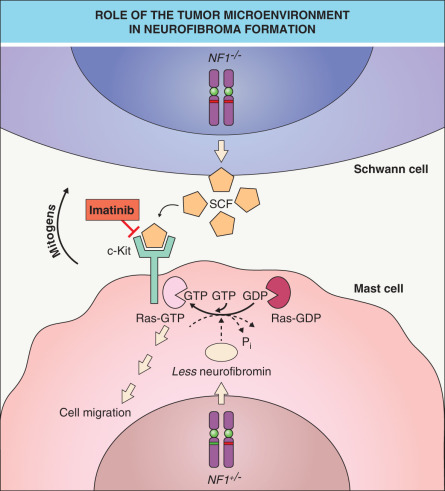
As observed in other autosomal dominant genodermatoses (e.g. basal cell nevus syndrome) caused by a defective tumor suppressor gene, a somatic “second hit” mutation inactivating the remaining allele of the gene (i.e. causing loss of heterozygosity [LOH]) can be found within skin lesions of NF1. Biallelic NF1 inactivation has been identified in Schwann cells from neurofibromas as well as melanocytes from café-au-lait macules in patients with NF1 . In a mouse model, Nf1 nullizygosity ( Nf1 −/− ) in Schwann cells was shown to be necessary, but not sufficient, for neurofibroma formation ; however, plexiform neurofibromas did arise from Nf1 −/− Schwann cells when the microenvironment included Nf1 +/− (rather than wild-type) mast cells . The latter cells appear early in neurofibroma development and are hypermotile in response to the c-Kit ligand (stem cell factor), which is oversecreted by Nf1 −/− Schwann cells. Once recruited, mast cells produce growth factors that likely promote neurofibroma tumorigenesis ( Fig. 61.2 ). Of note, genetic or pharmacologic attenuation of kit signaling in Nf1 +/− mast cells has been found to diminish neurofibroma initiation and progression (see Treatment section below).
Clinical Features
The clinical features of NF1 ( Table 61.2 ) are myriad and vary in frequency . Through decades of careful study, almost every organ system has been found to be involved in NF1. However, the cutaneous manifestations of NF1 represent particularly important clinical features and have a central role in its diagnosis .
| MAJOR CLINICAL FEATURES OF NEUROFIBROMATOSIS TYPE 1 |
| Cutaneous |
|
| Ocular |
|
| Skeletal |
| Cranial |
|
| Spinal |
|
| Limbs |
|
| Other |
|
| Tumors |
|
| Neurologic |
|
| Cardiovascular |
|
Oculocutaneous manifestations
Café-au-lait macules (CALMs) are tan to dark brown (depending on the degree of constitutive pigmentation), uniformly pigmented macules and patches that are randomly distributed on the body, with the exception of the scalp, palms, and soles. The CALMs of NF1 are sharply demarcated with regular borders and often have an oval shape ( Fig. 61.3 ); the diameter can vary from a few millimeters to >4 cm. CALMs may not be apparent at birth and frequently become more noticeable during the first year of life; fading of these lesions during later adulthood has also been described . When located within areas of dermal melanocytosis, CALMs may be surrounded by a rim of normally pigmented skin (see Ch. 112 ). Larger CALMs, especially those with associated hypertrichosis, should be palpated to exclude the possibility of an underlying plexiform neurofibroma (see below).
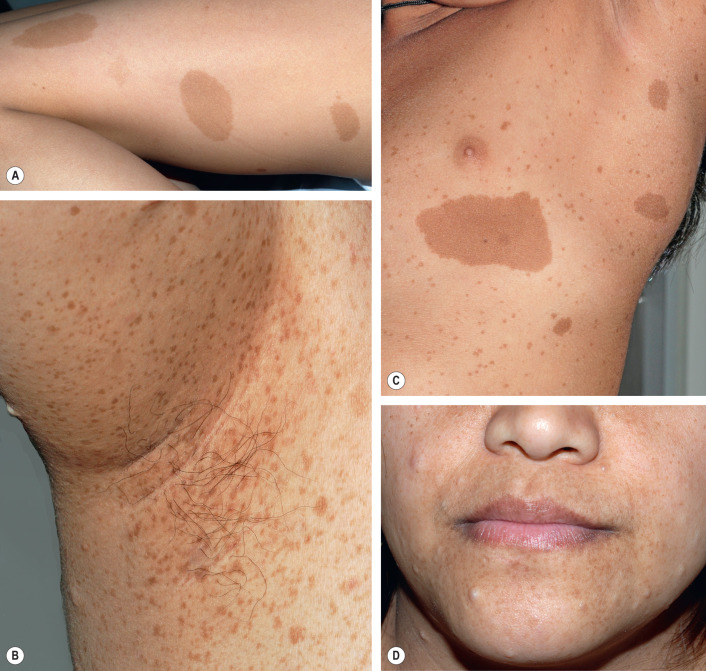
Crowe et al. reported that 10% of the normal population has one to five CALMs, and Burwell et al. found that 26% of 732 white schoolchildren had at least one CALM >1 cm in diameter. In the latter study, ≥6 CALMs were found in only three children (0.4%) and they were siblings with a parent who had NF1. For the clinical diagnosis of NF1, a minimum of six CALMs is required to meet the specificity of this criterion ( Tables 61.3 and 61.4 ). To avoid confusion with lentigines, CALMs must be >5 mm in prepubertal individuals and >15 mm in postpubertal individuals in order to satisfy this diagnostic criterion for NF1.
| NIH DIAGNOSTIC CRITERIA FOR NEUROFIBROMATOSIS TYPE 1 |
Two or more of the following must be present:
|
| DISORDERS ASSOCIATED WITH MULTIPLE CAFÉ-AU-LAIT MACULES | |
|---|---|
| Disorder | Notes |
| Neurofibromatosis and related disorders | |
| Neurofibromatosis 1 (NF1) | AD; six or more CALMs in >90% of patients; mutations in NF1 gene; see text |
| Neurofibromatosis 2 | AD; six or more CALMs in minority of patients; mutations in NF2 gene; see Table 61.6 |
| Mosaic (segmental) NF1 (formerly neurofibromatosis 5) | Neurofibromas alone or CALMs ± “freckling” in a block-like or other mosaic pattern; due to a postzygotic NF1 mutation, which may involve the gonads in some patients |
| Legius (NF1-like) syndrome | AD; six or more CALMs in >80% and intertriginous “freckling” in ~50% of patients; lack neurofibromas, Lisch nodules, optic gliomas, and typical osseous lesions of NF1; macrocephaly and learning disabilities are common; lipomas, hypopigmented macules, and vascular anomalies in minority; loss-of-function mutations in SPRED1 gene, the protein product of which inhibits MAPK signaling |
| Watson syndrome | AD; short stature; pulmonic stenosis; ID; may also have Lisch nodules, axillary/inguinal freckling, and neurofibromas; allelic with NF1; 60/100 * |
| Jaffe–Campanacci syndrome | Disseminated non-ossifying fibromas of long and jaw bones; hypogonadism or cryptorchidism; D; giant cell granulomas (jaws); usually due to NF1 mutation |
| Noonan syndrome | AD; short stature; pterygium colli; characteristic facies; cardiac defects, esp. pulmonic valve stenosis; skeletal and testicular abnormalities; keratosis pilaris atrophicans; lymphedema; melanocytic nevi; activating mutations in PTPN11 (allelic with LEOPARD syndrome) > SOS1, RAF1, RIT1 > KRAS, NRAS, MAP2K1, CBL, SHOC2 (with loose anagen hair), and rarely other genes; patients with an NF1–Noonan overlap phenotype have mutations in NF1 gene |
| Cardio-facio-cutaneous syndrome | AD; short stature; characteristic facies; low-set ears; cardiac defects; ID; sparse curly hair; dry skin to generalized ichthyosiform eruption; activating mutations in MAPK pathway genes – BRAF > MEK1, MEK2 > KRAS |
| Constitutional mismatch repair deficiency syndrome (childhood tumor syndrome with NF1 phenotype; formerly Turcot syndrome type 1) | AR; multiple CALMs, axillary freckling, neurofibromas and/or CNS gliomas (features similar to NF1) as well as hypopigmented macules, hematologic malignancies, and colorectal carcinoma; patients have mutations in both copies of a DNA mismatch repair gene, e.g. MLH1, MSH2, MSH6, PMS2 ; hereditary non-polyposis colorectal cancer (HNPCC) syndrome can occur in heterozygotes |
| McCune–Albright syndrome | Not inherited; presumably lethal unless in mosaic state; polyostotic fibrous dysplasia; hyperfunction of endocrine glands, esp. gonads (e.g. precocious puberty); CALMs usually appear during infancy and can overlie bony changes; CALMs classically end at midline, but so can larger CALMs of NF; CALMs may follow lines of Blaschko; activating mutations in the GNAS1 gene that encodes G s α; 35/100 * |
| Disorders with additional cutaneous findings | |
| Tuberous sclerosis complex (TSC) | AD; ID; seizures; hypopigmented macules; angiofibromas; fibrous plaques; mutations in TSC1 and TSC2 genes; see text |
| Westerhof syndrome | AD; congenital hypo- and hyperpigmented macules; ID; growth retardation |
| Piebaldism | AD; CALMs in involved as well as uninvolved skin; mutations in KIT proto-oncogene |
| Familial progressive hyper- and hypopigmentation | AD; CALMs of variable sizes admixed and overlapping with lentigines and hypopigmented macules and patches; activating mutations in KITLG gene |
| Mukamel syndrome | AR; premature graying (infancy); lentigines; depigmented macules; ID; spastic paraparesis, microcephaly, scoliosis |
| Noonan syndrome with multiple lentigines (LEOPARD syndrome) | AD; darker “café noir” macules; L entigines, E CG abnormalities, O cular hypertelorism, P ulmonic stenosis, A bnormal genitalia, R etardation of growth, D eafness syndrome; mutations in PTPN11 ; 38/100 * |
| Carney complex (NAME and LAMB syndromes) | AD; darker “café-noir” macules, lentigines, blue nevi; cutaneous and atrial myxomas; endocrine neoplasia of adrenal glands, pituitary gland, and/or testes; mutations in PRKAR1A |
| Partial unilateral lentiginosis | Agminated lentigines; debatable if associated neurologic or psychiatric problems; unilateral axillary freckling and Lisch nodules have been described (such patients likely have mosaic NF1) |
| Bannayan–Riley–Ruvalcaba syndrome ‡ | AD; penile lentigines; vascular malformations; lipomas; intestinal hamartomas; macrocephaly; mutations in PTEN gene; ≤10% of patients have CALMs |
| Cowden syndrome ‡ | AD; multiple tricholemmomas; cobblestoning of oral mucosa; palmoplantar keratoses; sclerotic fibromas, hamartomas and carcinoma of breast, thyroid, colon; mutations in PTEN gene; ≤10% of patients have CALMs |
| Gastrocutaneous syndrome † | AD; multiple lentigines; peptic ulcer/hiatal hernia; hypertelorism; myopia |
| Ataxia–telangiectasia | AR; telangiectasias of conjunctivae, head/neck, and acral sites; premature canities; cerebellar ataxia; abnormal DNA repair; lymphomas and leukemias; immunodeficiency; sinopulmonary infections; mutations in ATM gene; 20/100 * |
| Bloom syndrome | AR; telangiectatic erythema of face; photosensitivity; growth retardation; increased sister chromatid exchanges; malignancies; hypogonadism; mutations in BLM gene that encodes the RecQ protein-like-3 DNA helicase |
| Fanconi anemia | AR; generalized hyperpigmentation, esp. flexural ± guttate “hypopigmented” macules; pancytopenia; skeletal malformations, e.g. aplasia radii; chromosomal fragility and malignancies, e.g. acute myelogenous leukemia |
| Chromosomal anomalies/mosaicism | In particular, ring chromosomes (7, 11, 12, 15, 17); rearrangements of 7 and 14; ring 7 also associated with vascular lesions (hemangiomas and capillary malformations) and congenital melanocytic nevi |
| Extensive epidermal and sebaceous nevi | Mosaic; “syndrome” includes seizures, ID, musculoskeletal and ocular abnormalities; hypophosphatemic vitamin D-resistant rickets; see Table 62.7 for genes |
| Multiple endocrine neoplasia 1 | AD; tumors of the pituitary, parathyroid, and pancreatic islet cells; multiple facial angiofibromas, collagenomas, gingival papules, confetti-like hypomelanotic macules, lipomas; MEN1 gene |
| Multiple endocrine neoplasia 2B | AD; mucosal neuromas; marfanoid habitus; medullary thyroid carcinoma; intestinal ganglioneuromatosis; pheochromocytoma; mutations in RET proto-oncogene |
| Johnson–McMillin syndrome | Probable AD; alopecia; anosmia; deafness; hypogonadism; microtia |
| Tricho-hepato-enteric syndrome | AR; CALM and lentigines on hips and legs; diffuse pigmentary dilution (skin & hair), woolly hair; dysmorphic facies with hypertelorism; intractable diarrhea, liver disease; immune deficiency; mutations in TTC37 or SKIV2L |
| Tay syndrome † | ?AR; growth retardation; ID; triangular face; cirrhosis; trident hands; premature canities; vitiligo |
| Other disorders § | |
| Russell-Silver syndrome | Growth retardation; triangular face; clinodactyly 5th finger; hemihypertrophy; epigenetic changes on chromosome 15p13 or maternal uniparental disomy of chromosome 7; 45/100 * |
* Estimates of prevalence of CALMs .
† To date, observed in a single family.
‡ Overlapping conditions collectively referred to as PTEN hamartoma tumor syndrome.
§ Single case reports of multiple CALMs in association with X-linked hypohidrotic ectodermal dysplasia, woolly hair, and osteoma cutis.
In its fullest expression, NF1 can manifest with thousands of neurofibromas in an affected individual, hence the appellation neurofibromatosis ( Fig. 61.4 ). Several variants of neurofibromas are observed in NF1. Most commonly, proliferations of spindle cells occur within the dermis, leading to cutaneous neurofibromas (CNFs). CNFs are skin-colored to pink, tan, or brown papulonodules that are soft or slightly rubbery in texture and can range from a few millimeters to several centimeters in diameter. While fully formed CNFs are often dome-shaped or pedunculated, early lesions may be barely elevated. They invaginate easily with gentle pressure, exhibiting the pathognomonic “buttonhole” sign. Although usually asymptomatic, lesions may become pruritic or, occasionally, irritated. CNFs appear as early as 4–5 years of age but more typically develop around puberty, with occasional acceleration of growth during pregnancy.
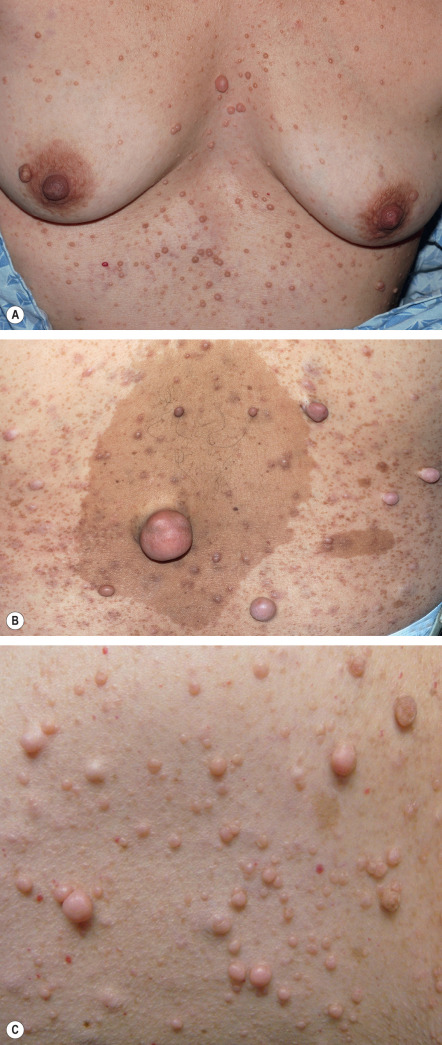
Subcutaneous neurofibromas (SNFs) occur deeper in the dermis and subcutis, tending to be firmer and less well-circumscribed than CNFs. Nearly one-fifth of NF1 patients have at least one SNF , and one-quarter of these individuals develop tumor-related local complications. Less common cutaneous presentations of neurofibromas in patients with NF1 include: (1) blue–red macules due to thick-walled blood vessels as well as neurofibromatous tissue within the papillary dermis ( Fig. 61.5 ); and (2) pseudo-atrophic macules due to replacement of collagen in the reticular dermis with neurofibromatous tissue .
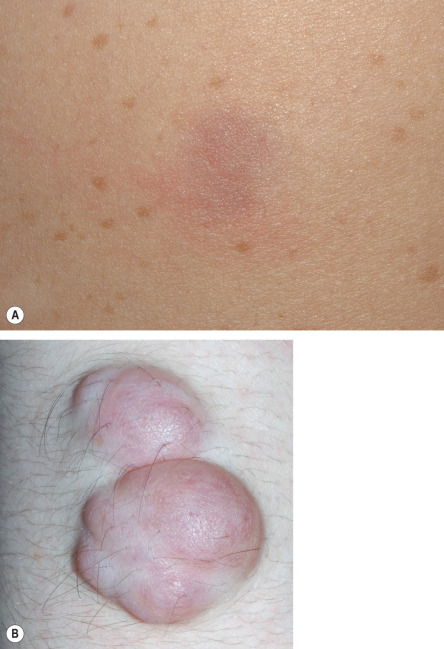
Plexiform neurofibromas (PNFs; Fig. 61.6 ) may track along nerves to create tender, firm nodules or masses in the subcutaneous tissue with a “bag of worms” consistency upon palpation, or they can infiltrate diffusely to involve various layers of the skin, fascia, muscle, or even internal structures. Approximately 25% of NF1 patients have clinically apparent PNFs. Larger lesions may cause soft tissue and bony hypertrophy, and massive distortion of the head and neck or extremities occasionally develops. Impingement upon nerves can lead to neurologic deficits.
Unlike other neurofibromas, PNFs are probably all congenital in origin. Superficial and/or diffuse lesions usually become clinically apparent by the age of 4 or 5 years , while deep/internal nodular lesions may remain undetected even in adulthood unless a full-body MRI is performed . Older children without evidence of soft tissue overgrowth related to a PNF are unlikely to develop this finding later in life. Both hyperpigmentation and hypertrichosis can overlie PNFs , sometimes leading to misdiagnosis as a congenital melanocytic nevus (see Fig. 61.6 ).
The transformation of PNFs into malignant peripheral nerve sheath tumors (MPNSTs, also known as “neurofibrosarcomas”) occurs in 3–15% of patients with NF1 , with a peak incidence in young adults. The onset of rapid growth, increased firmness, or persistent pain within an established PNF or the development of a new neurologic deficit may herald malignant degeneration ( Table 61.5 ; see Management section ). MPNSTs can be very aggressive and are often fatal .
| EVALUATION AND MANAGEMENT OF NEUROFIBROMATOSIS 1 (NF1) PATIENTS |
| At time of diagnosis and annually during childhood and adolescence (unless otherwise indicated) |
|
| Minimum annual evaluation for adults with uncomplicated disease |
|
| Potentially affected family members |
|
* Immediate assessment is required for any of these findings.
† The role of neuroimaging in asymptomatic patients is controversial.
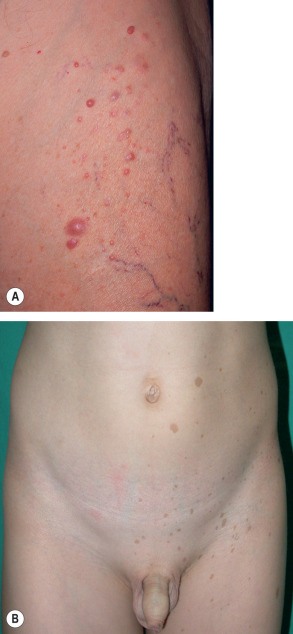
“Freckling” in the axillae and groin in NF1 patients was first noted by Crowe and is referred to as “Crowe’s sign” (see Fig. 61.3 ). Numerous 1–3 mm brown macules (more akin to lentigines than ephelides) typically populate these intertriginous zones, the neck, and, occasionally, the majority of the skin surface (see Figs 61.3 & 61.4 ). Involvement of the axillae is most common, occurring in about 80% of NF1 patients . The lesions usually arise by 4–6 years of age, following the emergence of CALMs but preceding the development of neurofibromas. As a result, intertriginous “freckling” is a useful clinical sign in the evaluation of a young child with multiple CALMs .
As many as 15–35% of children with NF1 develop one or more juvenile xanthogranulomas (JXGs) during the first 2–3 years of life, making these lesions a potential early sign of NF1 when they coexist with multiple CALMs . Young children with NF1 also have a 200- to 500-fold greater risk of developing juvenile myelomonocytic leukemia (JML; incidence 1/2000–1/5000) than does the general pediatric population. Although a “triple association” between NF1, JML, and JXG has been described , the magnitude of additional risk of JML in children with NF1 plus JXGs compared to NF1 alone remains to be determined. Neurofibromin is known to regulate the proliferation of myeloid cells, but the exact etiopathogenesis of the “triple association” remains unclear.
Vascular anomalies that have been recognized as manifestations of NF1 include nevus anemicus and glomus tumors . In two prospective studies, a nevus anemicus was identified in a total of 55% (92/166) of children and 36% (19/53) of adults with NF1, compared to only 2% (6/302) of age- and sex-matched controls . These lesions favor the trunk, especially the mid chest, and become more evident after rubbing the skin. Because nevus anemicus is prevalent in NF1 patients and apparent by early childhood, its use as a diagnostic criterion has been proposed .
Biallelic inactivation of NF1 has been documented within glomus tumors in NF1 patients . The tumors are often multiple and develop on the fingers > toes; like their sporadic counterpart, present with paroxysmal pain and sensitivity to cold (see Ch. 114 ). Focal swelling, ill-defined reddish discoloration (often of the nail bed), and nail dystrophy may be observed.
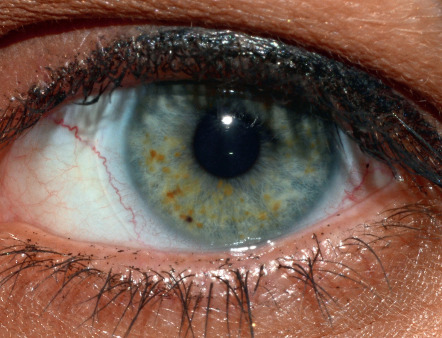
Pigmented iris hamartomas, or Lisch nodules, begin to develop at about 3 years of age and are found in ~90% of NF1 patients by 20 years of age . These 1–2 mm, dome-shaped, yellow–brown papules are best seen on slit-lamp examination ( Fig. 61.7 ). Lisch nodules rarely lead to symptoms or complications. Asymptomatic choroidal nodules are also present in 80–100% of adults and 60–70% of children with NF1, and their use as a diagnostic criterion has been suggested. However, the near-infrared reflectance imaging required for detection of these lesions may be technically challenging in younger children .
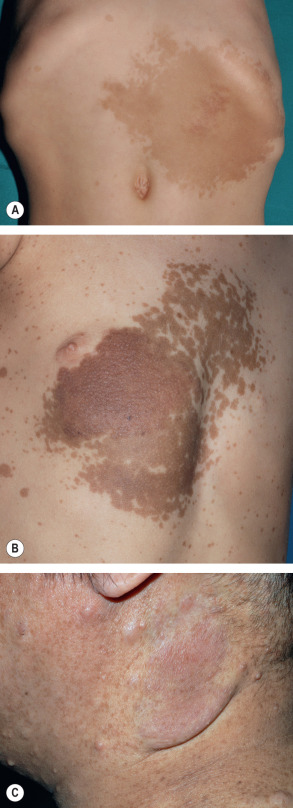
In mosaic (segmental) NF1 (formerly known as NF5), cutaneous neurofibromas, CALMs, and/or “freckling” are present in one or more dermatomes (especially for neurofibromas; Fig. 61.8A ) or in other mosaic distribution patterns such as blocks with midline demarcation (especially for CALMs and freckling; Fig. 61.8B ). This form of NF1 results from a postzygotic NF1 mutation, and up to 30% of patients also have extracutaneous manifestations of NF1 . If the mutation involves the gonads (germline mosaicism) as well as the skin, there is a chance of full-blown NF1 in the patient’s offspring.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


