Abstract
This chapter discusses the classification, terminology, and histogenesis of the most common cutaneous neural tumors along with their key clinical and pathologic features as well as differential diagnosis. These tumors can be classified into two major groups: those derived from peripheral nerves and those derived from ectopic/heterotopic neural tissue. Precise diagnosis is based on a combination of clinical presentation, histopathologic features, and immunohistochemical and/or molecular aspects. The vast majority of neural tumors are benign, but rare malignant variants may occur. Some neural tumors are associated with multisystem syndromes (e.g. neurofibromatosis, Carney complex). Although not a neural tumor, Merkel cell carcinoma is included in this chapter because it shares several structural and immunohistochemical features with neuroectodermally derived cells. The majority of Merkel cell carcinomas are associated with a specific polyoma virus, the Merkel cell polyomavirus, and they represent one of the most aggressive cutaneous malignancies.
Keywords
cutaneous neural tumors, peripheral nerve sheath neoplasms, heterotopic neural tissue, neuroma, schwannoma, neurofibroma, nerve sheath myxoma, cellular neurothekeoma, granular cell tumor, perineurioma, malignant peripheral nerve sheath tumor, Merkel cell carcinoma, heterotopic neuroglial tissue, heterotopic meningeal tissue, heterotopic neuroblastic tissue, neuroblastoma
In the past, neural tumors were often misdiagnosed histopathologically because of confusing classifications. As a result, their clinical relevance was poorly understood. Clinically, cutaneous neural tumors often look alike and most of them are benign. However, their correct diagnosis can be helpful in recognizing important clinical syndromes and this can contribute to better patient management .
Classification, Terminology, and Histogenesis
Cutaneous neural tumors can be classified into two major groups: those derived from peripheral nerves and those derived from ectopic or heterotopic neural tissue ( Table 115.1 ). The former group is often subdivided into true nerve sheath neoplasms and hamartomatous tumors, although this classification may not be universally accepted .
Cutaneous neural tumors either arise from or differentiate toward one or more elements of the nervous system. During their differentiation, neural neoplasms often recapitulate to varying degrees the morphogenesis of normal peripheral nerves. Therefore, knowledge of the organization of the normal peripheral nerve is crucial to understanding the histogenesis of tumors that arise from it . The peripheral nerve can be compared to a conventional telephone cable, in which each axon and its surrounding Schwann cell layer correspond to a telephone wire and its insulation, respectively ( Fig. 115.1 ).
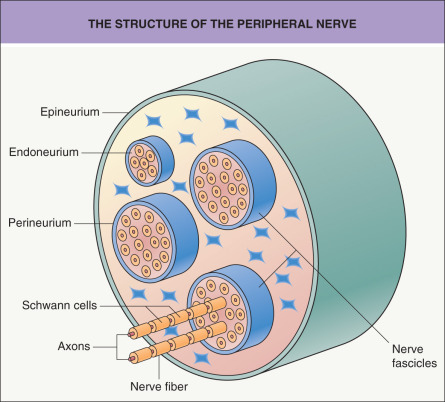
The basic units of a peripheral nerve are nerve fibers, composed of axons and the surrounding Schwann cells. These fibers form nerve fascicles and they are held together by a sheath of specialized cells, which is called the perineurium. The space between the individual nerve fibers is called the endoneurium. As in a telephone cable system where the smaller cable units are separated, protected and held together by an outer wrapping, bundles of nerve fascicles are also encased in a supportive fibrous sheath that is called the epineurium. To a variable extent, this architectural arrangement is recognizable in many cutaneous neural neoplasms.
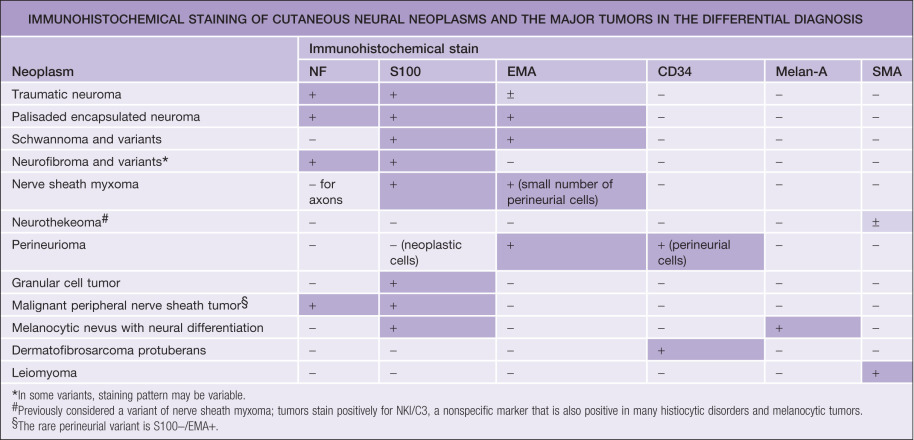
The most important constituent cells are the Schwann cell, the perineurial cell, and the various nonspecific mesenchymal cells, such as fibroblasts and mast cells. These cells are capable of proliferation and malignant transformation. Other elements of the peripheral nerve, which are cell parts or products (i.e. axons and myelin, respectively), cannot duplicate. Schwann cells are derived from the neural crest, and there is evidence that perineurial cells are modified fibroblasts of mesodermal origin. This difference in histogenesis is also reflected in the distinct antigenic expression of these cells. Schwann cells express S100 protein but not epithelial membrane antigen, whereas perineurial cells stain for epithelial membrane antigen but not for S100 protein. Axons contain a specific type of intermediate filament called neurofilament, and myelinated axons contain myelin basic protein, both of which can be detected by immunohistochemistry. These and other immunohistochemical markers often can help to establish the correct diagnosis.
In the following sections, each clinically relevant neural neoplasm is discussed ( Tables 115.2–115.4 ).
| CLINICAL FEATURES OF BENIGN NEURAL NEOPLASMS | |||||||
|---|---|---|---|---|---|---|---|
| Traumatic neuroma | Palisaded encapsulated neuroma (PEN) | Schwannoma (neurilemmoma) | Neurofibroma | Nerve sheath myxoma * | Cellular neurothekeoma * | Granular cell tumor | |
| Incidence | Uncommon | Rare | Uncommon | Very common | Rare | Rare | Rare |
| Age | Any | Adults (mean age, 45.5 years) | Adults (20–50 years) | Adults (20–60 years) | Adults (mean age, 48 years) | Early adulthood (mean age, 24 years) | Adults (30–50 years) |
| Gender | M : F = 1 : 1 | M : F = 1 : 1 | F > M | M : F = 1 : 1 | M : F = 1 : 2 | F > M | M : F = 1 : 3 |
| Number | Usually solitary | Usually solitary | Usually solitary (>90%) | Usually solitary | Usually solitary | Usually solitary | Usually solitary |
| Location | At sites of trauma, surgical scars, amputations | 90% face, 10% elsewhere | Flexor aspect of an extremity, head | Trunk, head (solitary type) | Head and upper extremities | Predominantly on head, but also elsewhere | 30% tongue, 70% elsewhere, mainly head and neck |
| Size | 0.5–2.0 cm | 0.2–0.6 cm | 0.5–3.0 cm | 0.2–2.0 cm | 0.5–1.0 cm | 0.5–3.0 cm | 0.5–3.0 cm |
| Clinical appearance | Skin-colored to reddish purple, firm papule or nodule | Skin-colored or pink, rubbery to firm papule or nodule | Soft, skin-colored to pink or yellow, smooth-surfaced nodule or tumor | Skin-colored, soft or rubbery papule or nodule, sometimes pedunculated | Soft, skin-colored papule or nodule | Pink, red or brown, firm papule or nodule | Skin-colored to brownish red, firm papulonodule; may have ulcerated or verrucous surface |
| Symptoms | Variable; tingling, pruritus, lancinating pain | Asymptomatic | Asymptomatic; occasionally painful or tender; rarely paresthesias; occasionally freely movable | Asymptomatic; “buttonhole” sign may be present | Asymptomatic | Usually asymptomatic, rarely painful or pruritic | Asymptomatic or occasionally tender or pruritic |
| Association | Multiple neuromas in MEN2B (mucosal) and in PTEN hamartoma syndrome (mucocutaneous) may resemble traumatic neuromas | Multiple (non-encapsulated) neuromas in MEN 2B (mucosal) and PTEN hamartoma tumor syndrome (mucocutaneous) | Neurofibromatosis (primarily type 2) | If multiple, may be sign of one of the various forms of neurofibromatosis (see Ch. 61 ) | NA | NA | 10% multiple; predilection for blacks; rare in children |
| Clinical differential diagnosis | Hypertrophic scar, dermatofibroma, foreign body granuloma, granular cell tumor, neurothekeoma | Basal cell carcinoma, adnexal tumor, IDN, neurofibroma, schwannoma, nerve sheath myxoma; when multiple, adnexal tumors (see Fig. 111.5 ) | Neurofibroma, lipoma, angiolipoma, cyst (epidermoid, pilar, dermoid, ganglion), IDN, adnexal tumors (including sebaceous), PEN, leiomyoma | IDN, soft fibroma, schwannoma, fibrolipoma, PEN, nerve sheath myxoma, anetoderma, lipoma, cyst (epidermoid, pilar) | IDN, neurofibroma, schwannoma, lipoma, cyst (myxoid, ganglion, epidermoid, pilar) | Dermatofibroma (including sclerosing hemangioma variant), adnexal tumor, hypertrophic scar, keloid, granular cell tumor, PEN, fibrosed dermal or compound melanocytic nevus | Tongue: irritation (traumatic) fibroma, verruca, SCC Elsewhere: dermatofibroma, regressing verruca, adnexal tumor, prurigo nodularis, fibrosed melanocytic nevus, SCC |
| Other | “Rudimentary supernumerary digit” is considered a variant ( Ch. 64 ) | May be induced by minor trauma | May be multiple; “schwannomatosis” | Plexiform variant is pathognomonic for neurofibromatosis type 1 | Visceral forms occur; malignant transformation may occur (3%) | ||
| HISTOLOGIC DIFFERENTIAL DIAGNOSTIC FEATURES OF COMMON CUTANEOUS NEURAL NEOPLASMS | ||||
|---|---|---|---|---|
| Traumatic neuroma | Palisaded encapsulated neuroma | Schwannoma | Neurofibroma | |
| Location in the skin | Any level of dermis or subcutis | Mid dermis, occasionally extending into subcutis | Deep dermis or subcutis | Any location in the dermis |
| Growth pattern | Usually well circumscribed but may be poorly defined at the distal end | Well circumscribed, nodular, rarely plexiform | Well circumscribed, nodular or ovoid | Poorly to well circumscribed |
| Encapsulation | Usually encased by a fibrous sheath | Yes, by perineurium | Yes, by perineurium | Not encapsulated in the dermis |
| Architecture | Chaotic, poorly organized tangle of fascicles of various sizes and shapes | Compactly arranged fascicles; frequent clefts between fascicles | Hypercellular areas (Antoni A), fascicles in various patterns, hypocellular areas (Antoni B), edematous, myxoid | Fine fibrillary lattice of haphazardly arranged spindle cells in variably dense matrix |
| Constituent cell types | Schwann cells, fibroblasts, perineurial cells, inflammatory cells, macrophages | Schwann cells (99%), perineurial cells (in capsule only) | Schwann cells (99%), perineurial cells (in capsule only) | Schwann cells, perineurial cells, fibroblasts, mast cells |
| Cytologic features | Spindle cells with indistinct cytoplasmic contours and tapered slender nuclei | Spindle cells with indistinct cytoplasmic contours and tapered slender nuclei | Spindle cells with indistinct cytoplasmic contours and slender nuclei | Spindle cells with slender nuclei, plump fibroblasts |
| Nuclear palisading | None | Usually present, but indistinct | Yes, prominent in Antoni A areas | Rarely |
| Verocay bodies | None | None | Often | None |
| Nerve fibers (axons) | Yes; abundant, irregular pattern | Yes; abundant, often in a parallel arrangement | None or only at the site of the connecting nerve | Yes; rare, scattered |
| Other important features | Nerve of origin frequently present, extensive fibrosis | Nerve of origin frequently present, no fibrosis | Mast cells; may have extensive degenerative changes (e.g. hyalinization, hemorrhage) in Antoni B areas (“ancient changes”) | Variable fibrosis and myxoid changes, mast cells, occasional blood vessels |
| Histopathologic differential diagnosis | Hypertrophic scar, neurofibroma, schwannoma, palisaded encapsulated neuroma | Traumatic neuroma, schwannoma, angioleiomyoma, myofibroma | Palisaded encapsulated neuroma, angiomyoma, fibrous histiocytoma, traumatic neuroma | Melanocytic nevus with neuroid differentiation, dermatofibroma, hypertrophic scar, dermatofibrosarcoma protuberans, traumatic neuroma |
Neuromas
Traumatic neuroma
- ▪
At the site of previous trauma
- ▪
Skin-colored papule or nodule
- ▪
Often painful or sensitive
- ▪
Circumscribed histologically, but not encapsulated
- ▪
Regenerative proliferation of axons and Schwann cells plus fibrous tissue
Palisaded encapsulated neuroma (PEN)
- ▪
Solitary, often skin-colored papule or nodule
- ▪
Usually on the face of adults
- ▪
Skin-colored papule or nodule
- ▪
Circumscribed histologically; partially or completely encapsulated
- ▪
Compactly arranged fascicles of spindle cells with vague palisading of nuclei
Introduction
Neuromas are proliferations of neural tissue in which Schwann cells and axon components are found in roughly equal numbers. There are two major subtypes: traumatic or amputation neuroma and palisaded encapsulated neuroma (PEN). Traumatic or amputation neuromas are complex regenerative proliferations of nerve fibers secondary to injury. Palisaded encapsulated or solitary circumscribed neuromas are complex proliferations of nerve fibers without apparent previous tissue injury.
History
The current view of traumatic neuroma was introduced by Huber and Lewis based on the concept of Wallerian degeneration. PEN was described by Reed and co-workers in 1972 .
Epidemiology
Traumatic neuromas are relatively uncommon but can occur at any age and in either gender. They are more prevalent in professions with a high probability of physical injuries. The solitary form of PEN develops spontaneously and gradually without evidence of obvious previous trauma. These tumors appear during adulthood (mean age, 45.5 years) with nearly equal occurrence in men and women . With the solitary form, there is no specific association with neurofibromatosis or multiple endocrine neoplasia syndrome type 2B (MEN 2B).
Pathogenesis
Any extrinsic damage to nerve fibers can cause a traumatic neuroma. Amputation neuroma is considered the most common form and represents an attempted, but failed, regeneration of nerve fibers following transection. After transection, the distal segments of the nerve fibers degenerate, whereas the proximal segments regenerate in an attempt to reunite with the distal portion of the transected nerve fibers . In cases of severe trauma, this regenerative process is unsuccessful and the growing nerve fibers form a tangle of fascicles within fibrotic tissue. Despite marked variations in arrangement, size and shape of the regenerating fascicles, in traumatic neuromas the constituent fibers have a Schwann cell to axon ratio close to normal (1 : 1), which helps to distinguish true neuromas from other nerve sheath neoplasms .
In PEN there is an overgrowth of axons and their sheath cells within the confinement of the perineurium. This benign tumor most likely represents a hamartomatous growth in which there is a close reduplication of the normal axon to Schwann cell ratio . The cause of the overgrowth of neurites is unknown. Although minor tissue injury such as inflammation induced by acne has been suggested as a cause, to date a definite traumatic origin has not been established .
Clinical Features
Traumatic neuromas are usually solitary, skin-colored or reddish purple, firm papules or nodules at sites of wounds, surgical scars, and amputations ( Fig. 115.2 ). On the lower extremities they tend to be multiple . Early lesions are asymptomatic, but after a few months they gradually become painful, frequently with a lancinating character. Variable tingling and itching can be associated with the pain.
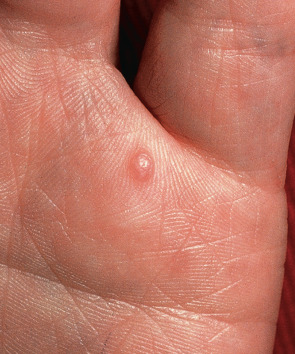
In neonates and young infants, lesions may be located on the lateral surface of a normal digit, most commonly the ulnar side of the fifth digit. Here, they represent amputation neuromas secondary to amputation in utero of supernumerary digits (see Ch. 64 ). These tumors are occasionally referred to as “rudimentary supernumerary digits”; however, on histologic examination, they contain neither normal nor rudimentary elements of a digit.
In the solitary form of PEN, the individual lesions are asymptomatic, rubbery to firm, skin-colored to pink papules or nodules ranging in diameter from 0.2 to 0.6 cm . Approximately 90% are located on the face, primarily around the nose, but they also occur on the cheek, chin and lips . The remaining 10% occur elsewhere, including the trunk and extremities.
In MEN 2B, also referred to as multiple mucosal neuroma syndrome or MEN type 3, numerous soft, skin- or mucosa-colored papules and nodules appear on the lips, tongue and conjunctivae, as well as in the nasal and laryngeal mucosa. In addition to rare neuromas of perinasal skin, patients also develop pheochromocytomas, medullary carcinoma of the thyroid, gastrointestinal ganglioneuromas, and cutaneous hyperneuria (see Table 63.2 ); the latter is characterized by an increased number of hyperplastic dermal nerves, but without the nodular configuration seen in neuromas. Multiple mucocutaneous neuromas with a predilection for the face and distal extremities can also be a manifestation of the PTEN hamartoma tumor syndrome, which includes Cowden disease and Bannayan–Riley–Ruvalcaba syndrome (see Tables 63.3 & 63.4 ).
Pathology
Traumatic or amputation neuromas are usually well-circumscribed nodules located at any level of the dermis or the subcutis. They are encased by a fibrous sheath, although the distal end can be poorly defined. The proliferation is composed of a chaotic, poorly organized tangle of fascicles of various sizes and shapes ( Fig. 115.3 ). Between the fibers are variable amounts of fibrous tissue with or without inflammatory cells or mucin . The constituent cells are Schwann cells and perineurial cells with spindle-shaped nuclei and cytoplasm ( Fig. 115.4 ). Special stains show many axons in an irregular, haphazard pattern .
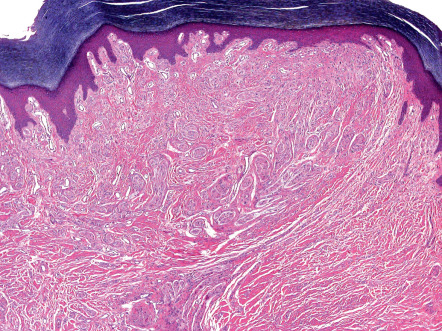
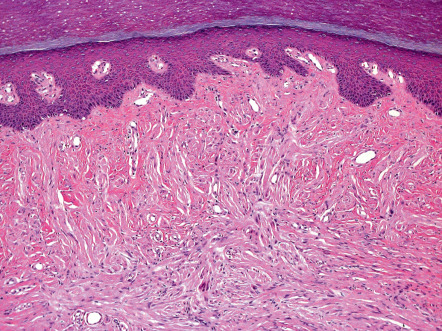
Solitary PENs are well-circumscribed, ovoid or round tumors located in the mid dermis, although some lesions may extend into the subcutis. The tumor appears encapsulated by a thick condensation of collagen fibers that surround it, and there is often some clefting from the adjacent dermis ( Fig. 115.5 ). The parenchyma is composed of interwoven fascicles of spindled cells. The fascicles are compactly and relatively uniformly arranged, separated only by clefts. There is no evidence of extensive fibrosis, inflammation, granulation tissue, degenerative changes or foreign bodies, in contrast to traumatic neuromas .
The nuclei of the spindle-shaped tumor cells are elongated and wavy with tapered ends, and they have an evenly basophilic chromatin pattern. Occasionally, a parallel arrangement of the nuclei is present, but, despite its name, distinct palisading or Verocay body formation is rare . There is no appreciable nuclear pleomorphism, and mitotic figures are scant or absent. Special stains show abundant axons in a variable pattern . The tumors of MEN 2B share some features with PEN but are not encapsulated and are multiple.
Epithelial sheath neuroma represents a more recently described variant of neuroma in which nerve fascicles are present in the superficial dermis and each fascicle is surrounded by an epithelial sheath. This neuroma variant must be distinguished from perineural invasion by malignant tumors .
Differential Diagnosis
The diagnosis of traumatic neuroma is usually suspected by the history of a painful or symptomatic papulonodule at a site of injury and is confirmed by pathologic examination.
A PEN can resemble a BCC, adnexal tumor, intradermal melanocytic nevus, or neurofibroma (see Table 115.2 ). The diagnosis is seldom made clinically and depends upon histologic examination (see Table 115.3 ).
Treatment
Surgical excision of traumatic neuromas is recommended, with an attempt to reposition the proximal nerve stump into a scar-free area . PENs are easily excised, and some can even be enucleated from the surrounding dermis or subcutaneous tissue .
Schwannoma
▪ Neurilemmoma ▪ Neurolemmoma ▪ Schwann cell tumor ▪ Acoustic neuroma
- ▪
Solitary papulonodule that usually appears on the flexural aspect of an extremity, along a peripheral nerve
- ▪
Occasionally painful or tender
- ▪
Deep dermal or subcutaneous location
- ▪
Histologically, there is an encapsulated spindle cell proliferation with biphasic Antoni A and B areas, palisading of nuclei, and Verocay bodies; axons are usually absent
Introduction
Schwannomas are benign true nerve sheath neoplasms composed entirely of a Schwann cell proliferation.
History
Stout coined the term “neurilemoma”, referring to the closely applied sheath and covering of the tumor . Because nearly the entire tumor is composed of Schwann cells, the designation “schwannoma” is now preferred.
Epidemiology
Schwannomas are relatively uncommon tumors that can occur at any age, but they occur most often in adults and slightly more frequently in women than in men. Approximately 90% of schwannomas are solitary and not associated with any specific syndrome . That said, schwannomas are seen in patients with neurofibromatosis (primarily type 2) and therefore can be associated with bilateral acoustic neuromas and meningiomas (see Table 61.5 ) . Rare cases of multiple schwannomas have been referred to as “neurilemmomatosis” or “schwannomatosis”; sometimes there is a familial occurrence or features that overlap with neurofibromatosis type 2 (NF2) . Germline mutations in SMARCB1 have been described in familial schwannomatosis .
Pathogenesis
Schwannomas are derived from a proliferation of periaxonal or endoneurial Schwann cells. As a result of the Schwann cell proliferation, the remaining normal nerve fibers are displaced to the periphery of the tumor, creating an absence of the other components of a peripheral nerve . Only after careful dissection can the attached nerve trunk be demonstrated. Since only the Schwann cells proliferate, the neoplasm remains within the confines of the perineurium, forming an encapsulated tumor. The exact cause of the exclusive Schwann cell proliferation is unknown, but in some patients it is related to mutations in both alleles of NF2 . How changes in chromatin remodeling due to SMARCB1 mutations lead to schwannomatosis is also unclear.
Clinical Features
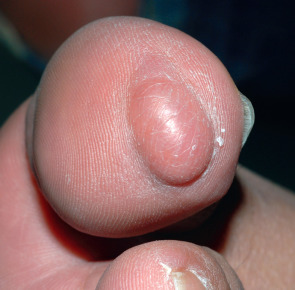
Schwannomas present as a solitary, soft, skin-colored to pink or yellow, smooth-surfaced, dermal or subcutaneous papulonodule ( Fig. 115.6 ). In general, their diameter ranges between 0.5 and 3.0 cm . They are most commonly found on the flexor aspect of an extremity along a larger nerve trunk, followed by the head and neck region . The tumors are usually asymptomatic, but occasionally pain and tenderness occur, in particular if the tumor is forcefully moved and the attached nerve is placed under tension. Motor disturbances and paresthesias are extremely rare .
Psammomatous melanocytic schwannoma is a rare variant that arises from spinal and autonomic nerves. Notably, in approximately half of the patients with this tumor, it represents a cutaneous manifestation of Carney complex (see Ch. 112 ). They present as well-circumscribed, brown–black to blue–gray papulonodules.
Pathology
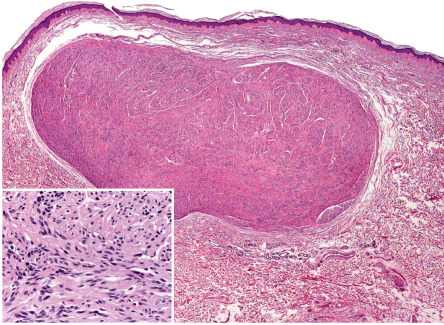
Schwannomas are well-circumscribed, nodular or ovoid tumors located in the deep dermis or subcutis. They are almost always encapsulated. The tumors are composed of hypercellular (also called Antoni A-type tissue) and hypocellular (Antoni B-type tissue) areas . The hypercellular areas show proliferation of spindle cells with indistinct cytoplasmic contours and uniform nuclei . Nuclear palisading and arrangement of palisaded nuclei in double rows, so-called “Verocay bodies”, are characteristic features of these tumors ( Fig. 115.7 ). Mitotic figures are absent or rare.
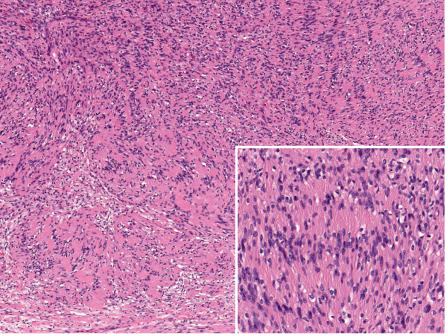
The hypocellular areas show variable degrees of degeneration, including cystic, edematous, mucinous, fibrotic, and vascular changes. Degenerative changes are often associated with some degree of cytologic atypia. These so-called ancient schwannomas should not be confused with the more specific entity of cellular schwannomas, which may display similar degenerative changes but rarely occur in the skin . As a general rule, schwannomas are devoid of axons or the latter can only be detected at the site at which the tumors are attached to a nerve .
There are additional less common subtypes of schwannomas, e.g. plexiform (no association with neurofibromatosis), pigmented, epithelioid, and glandular . In the psammomatous melanocytic schwannoma, a mixture of spindled and epithelioid cells that resembles syncytial tissue is seen. The cells have variable pleomorphism and nuclear atypia, and focal areas may demonstrate typical neural organization (e.g. nuclear palisading, Verocay bodies, whorl formation). Melanin deposition is often heavy, obscuring cytologic details. Psammomatous structures, i.e. eosinophilic laminated and whorled structures with variable calcification, are almost universally present. The cells stain positively for S100 protein, HMB-45, Melan-A, and synaptophysin .
Differential Diagnosis
The clinical differential diagnosis includes neurofibroma, lipoma, angiolipoma, adnexal tumor, cyst (epidermoid, pilar, dermoid, ganglion), and intradermal melanocytic nevus. The diagnosis is made by histopathologic examination. Clinically, a psammomatous melanocytic schwannoma may resemble a blue, combined, or atypical melanocytic nevus. Histologically, hybrid tumors with a combination of features of schwannoma and neurofibroma (or occasionally other benign neural tumors) have been described.
Treatment
Schwannomas are benign and a simple excision is curative. If the clinical diagnosis is suspected, the tumor should be removed by enucleation, preserving normal nerve function.
Neurofibroma
▪ Solitary neurofibroma – sporadic neurofibroma, solitary nerve sheath tumor ▪ Deep neurofibroma – subcutaneous neurofibroma ▪ Plexiform neurofibroma
Solitary type
- ▪
Skin-colored to tan–violet papule or nodule
- ▪
May be pedunculated or have the “buttonhole” sign
- ▪
Predilection for the trunk and head
- ▪
Spindle cell proliferation with variable mixture of the components of the peripheral nerve, including residual axons
- ▪
Fibrosis and mucinous changes are common
Plexiform type
- ▪
Pathognomonic of NF1
- ▪
Large, occasionally pigmented, bag-like mass
- ▪
Favors the trunk and proximal extremities
- ▪
The plexiform variant has similar cytologic constituents as the solitary or sporadic type, but carries a risk of malignant transformation
Introduction
Neurofibromas are benign tumors composed of a complex proliferation of neuromesenchymal tissue (Schwann cells, perineurial cells, fibroblasts and mast cells), with residual nerve fibers (axons) . Cutaneous neurofibromas are common, especially as solitary lesions. The presence of multiple neurofibromas raises the possibility of one of the several types of neurofibromatosis (including the mosaic form; see Ch. 61 ). Plexiform neurofibromas are seen in patients with neurofibromatosis type 1 (NF1).
History
Neurofibromas were originally described as fibrous tumors by von Recklinghausen. Subsequently, a neuroectodermal origin was suggested by Verocay, which was eventually confirmed by immunohistochemical and ultrastructural studies.
Epidemiology
Solitary cutaneous neurofibromas are relatively common, especially in adults, and have no gender preference. The presence of a plexiform neurofibroma almost always indicates that the patient has NF1.
Pathogenesis
Neurofibromas differ from schwannomas in that several cell types are involved in their histogenesis. Regardless of the various histologic subtypes of neurofibromas (e.g. solitary, diffuse, combined, plexiform), the basic process is proliferation of the entire “neuromesenchyme”, which includes Schwann cells, endoneurial fibroblasts, perineurial cells, mast cells, and cell types with intermediate features . Since the extent of proliferation of each cell line often differs, the resultant histologic composition and architecture will vary. As stated earlier, axons do not duplicate; therefore, the relative ratio of axons to Schwann cells will be less than 1 : 1. Studies in patients with NF1 indicate that their mutations lead to inactivation of one copy of NF1 (i.e. haploinsufficiency); however, tumorigenesis requires a somatic mutation that inactivates the remaining allele, i.e. a “second hit” leading to loss of heterozygosity (see Ch. 54 ). While nullizygosity ( NF −/− ) of Schwann cells is necessary, it may not be sufficient for neurofibroma formation; the latter may require an NF +/− environment, in particular NF +/− mast cells (see Ch. 61 ). The pathogenesis of isolated solitary neurofibromas is unclear.
Clinical Features
Neurofibromas are usually solitary, skin-colored, soft or rubbery papulonodules. They range in size from 0.2 to 2.0 cm and may become pedunculated . These tumors grow slowly and are asymptomatic. The “buttonhole” sign is often present , i.e. the tumor is easily invaginated. Approximately 10% of patients with common neurofibromas will have multiple lesions, and some of these patients have one of the several types of neurofibromatosis (see Ch. 61 ). There are several clinical and pathologic subtypes of neurofibroma, such as diffuse, pigmented, and plexiform variants. The diffuse and pigmented subtypes may present as indurated or soft nodules and plaques, with or without hyperpigmentation. Except for the plexiform variant, these subtypes of neurofibromas rarely undergo malignant change.
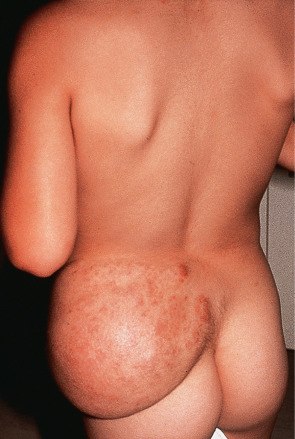
The plexiform neurofibroma is especially important because it is considered pathognomonic of NF1 (von Recklinghausen disease) and carries a higher probability of malignant transformation . Typically, these tumors are baggy or pedunculated, dermal and subcutaneous masses that can feel rope-like on palpation and are sometimes hyperpigmented ( Fig. 115.8 ). Recognition and correct patient management is crucial in these cases, especially in subtle forms of NF1, where the other cutaneous stigmata may not be obvious . The exact incidence of malignant transformation of plexiform neurofibromas is not known; it ranges from 2% to 13%, with the lower percentage cited more recently.
Pathology
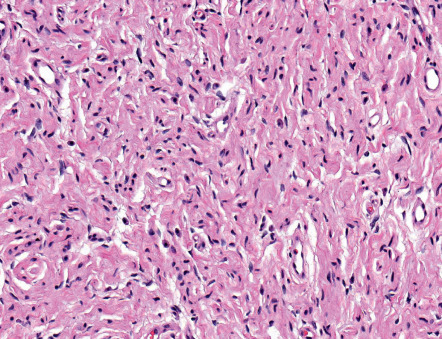
Neurofibromas are poor- to well-circumscribed, usually non-encapsulated, nodular or oblong tumors that can be located anywhere within the dermis and subcutis . The common solitary variant is usually present in the superficial dermis, creating a dome-shaped or polypoid elevation of its surface. The tumor is composed of a fine fibrillary lattice of haphazardly arranged slender spindle cells ( Fig. 115.9 ). The stroma can be variably vascular, fibrotic, edematous or myxomatous. In addition to Schwann cells and perineurial cells, plump fibroblasts and mast cells are present. Palisading of nuclei can occur, but true Verocay body formation is rare . Mitotic figures are absent or rare. The diffuse form has an essentially similar cytology, but lacks a well-defined growth pattern as it infiltrates adjacent structures. Atypical forms of neurofibromas have been described .
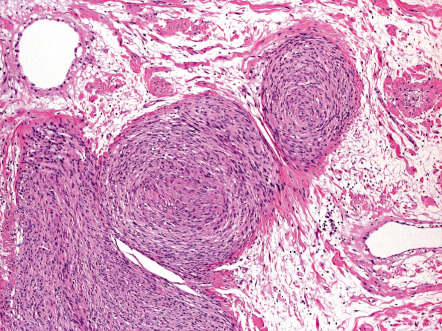
As opposed to superficial, solitary and diffuse forms, neurofibromas which are seated within the lower reticular dermis, subcutis, and other deep soft tissues are usually encapsulated by perineurium or epineurium and may show a plexiform growth pattern ( Fig. 115.10 ) ; the latter is defined as irregularly expanded, twisted, and interconnected nerve fascicles arranged in lobular structures. Variants with a combined diffuse and plexiform growth pattern usually occur in NF1. By utilizing special stains, rare scattered axons can be demonstrated in both the superficial and deep variants of neurofibroma.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


