A mutation is an event that produces heritable changes in the DNA. There are many different types of mutations, including point mutations (changes that imply loss, duplication, or alterations of small DNA segments, often involving a single or a few nucleotides) and major DNA changes (loss, duplication, or rearrangements of entire genes or of gene segments). This article reviews how different types of mutation may result in defective gene expression.
A mutation is an event that produces heritable changes in the DNA. There are many different types of mutations, including point mutations (changes that imply loss, duplication, or alterations of small DNA segments, often involving a single or a few nucleotides) and major DNA changes (loss, duplication, or rearrangements of entire genes or of gene segments). Each of these alterations is able to cause diseases. Simple patterns of mutation inheritance are observed in Mendelian diseases, that is when a single mutated gene is involved. The mode of inheritance in Mendelian conditions, such as all inherited forms of epidermolysis bullosa (EB), depends on whether a clinical phenotype is observed in the heterozygous carrier of the mutation (dominant inheritance) or only in the homozygote subjects born of healthy parents, who are heterozygous carriers of the mutation (recessive inheritance). Both recessive and dominant inheritance may be seen for mutant genes mapping on X sex chromosome (X-linked inheritance) and on autosomes (ie, nonsex chromosomes [autosomal inheritance]). Specifically, all genes involved in EB map on autosomes. The simplex (EBS) and dystrophic (DEB) EB types comprise variants with either dominant or recessive mode of inheritance; Kindler syndrome is a recessive condition, and junctional EB (JEB) is almost always recessively inherited.
The pattern of inheritance can be deduced on the basis of the family history; a pedigree (ie, a graphical diagram illustrating how the disease segregates in family members) can be drawn. In the case of an autosomal-dominant condition, the pedigree will show the transmission of the trait from generation to generation, usually without skipping (vertical transmission) and with several affected members, while the trait will be rare in autosomal-recessive conditions, presenting in one or more siblings who have healthy parents and offspring. Recessive inheritance should be suspected in consanguineous kindreds.
In exceptional cases, a recessive condition can arise from reduction to homozygosity of a mutation carried by a single parent. This phenomenon results from uniparental disomy (UPD, ie, the paradoxic inheritance of two copies of a mutant allele from one parent only). UPD is the result of events that operate to restore the correct chromosomal number in the embryo. Indeed, this number may become altered because of errors during meiosis and gamete maturation. UPD has been documented as the cause of EB in several cases.
Not all mutations are inherited. In some cases, a mutation can originate as a new event in the patient who will be a sporadic case of a dominant condition. However, a new mutation, also may involve germ cells in the gonad of a proband’s parent who, therefore, is a healthy mosaic for the disease allele. In this instance, an increased recurrence risk for what appears to be a new dominant mutation may occur; two affected children demonstrate the presence of germline mosaicism in one of the parents. In rare cases, specific types of new mutations also can result, when combined with a second inherited mutation, in a recessive condition. This can lead to diagnostic dilemmas in sporadic patients affected with diseases, such as DEB, which can be transmitted in either a dominant or recessive manner. Finally, a spontaneous mutation may occur postnatally within a somatic cell of an affected patient. If this event takes place in an EB individual and leads to the correction of the primary defect caused by the inherited mutation (genetic reversion), then the patient shows revertant somatic mosaicism, which manifests clinically as small patches of healthy nonblistering skin where blistering is no longer inducible. Such phenotypic rescue is caused by the expansion of the reverted somatic cells and might explain some atypical phenotypes that are milder than expected. Somatic reversion events are not so rare and have been documented extensively in JEB.
Mutations may alter inherited traits by affecting gene transcription, pre-mRNA splicing or protein translation, thus impacting on the expression or function of the protein product.
This article reviews how different types of mutation may result in defective gene expression.
Point mutations
Missense Mutations
Point mutations occur with the highest frequency in the human genome. If codons are changed by a single nucleotide substitution, these point mutations are defined as missense mutations. They directly involve crucial residues, such as at a catalytic site, or can alter the three-dimensional structure of the protein such that its function is compromised. Alternatively, they can perturb the correct assembly of protein subunits. An EB subtype that has clearly been shown to be caused by many different missense mutations affecting a specific protein domain is dominant dystrophic EB (DDEB). Such mutations lead to glycine substitutions in the triple helix of type VII collagen encoded by the COL7A1 gene. Type VII collagen is a trimer of identical chains [α1(VII) polypeptides]. Each chain is composed by a central large collagenous domain flanked by N- and C-terminal noncollagenous domains. The primary structure of the collagenous domain is remarkable, being formed, for the most of its length, by regular repeats of three amino acids, in which nearly every third residue is glycine (X-Y-Gly). Glycine is a key amino acid in this structure, because it occupies very little space and thereby allows the collagenous domain of three separate chains to come together and form a triple-stranded helical rod. Thus, a glycine substitution mutation in the α1(VII) polypeptide has a deleterious effect, because it becomes part of a triple-stranded structure, which will be partly unfolded, resulting in impaired collagen VII secretion into the extracellular matrix. From a genetic point of view, the allele with the missense mutation is dominant over the normal allele. It should be kept in mind, however, that not all glycine substitutions in the triple helix of type VII collagen result in dominant inheritance, as several of these missense mutations behave in a recessive manner.
Nonsense Mutations
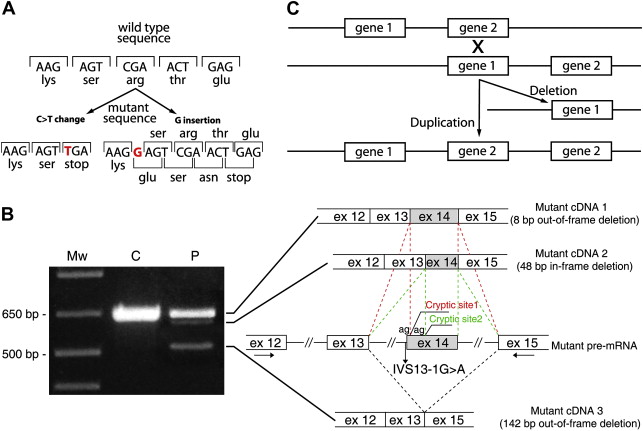
This type of point mutation affects protein translation via a nucleotide substitution that produces a premature termination codon (PTC) ( Fig. 1 ). PTC formation results in truncated proteins, which are usually unstable and degraded. PTC upstream of the penultimate exon, however, triggers a mechanism that leads to rapid degradation of the mutant transcript. This active process, called nonsense mediated mRNA decay (NMD), is evolutionary conserved and provides surveillance against faulty open reading frames (ORFs) by eliminating imperfect messages that contain PTCs and code for nonfunctional and potentially harmful polypeptides. For these reasons, nonsense mutations result in recessive inheritance and are normally associated, when present in both alleles, with the most severe variants of recessive DEB (RDEB) and JEB, which are characterized by a complete loss of expression of the protein product in the skin.
Frameshift Mutations
These are either deletions or insertions of one or a few nucleotides in the coding region of the gene (see Fig. 1 ). These mutations change the triplet code for all codons that follow and thus completely alter the downstream protein sequence, often leading to PTC formation. Therefore, they are genetically and functionally similar to the nonsense mutations.
Splicing Mutations
RNA splicing is crucial to normal gene expression, because introns must be excised precisely from the pre-mRNA to produce a mature mRNA for translation into protein. This process is performed by the spliceosome, a macromolecular apparatus endowed with the catalytic functions needed to join exons, and requires conserved sequence motifs at the intron–exon borders. These sequences are known as splice sites. The 5′ exon/intron junction is called the donor splice site, and the 3′ intron/exon junction is the acceptor splice site. The 5′ end of the intron almost invariably begins with nucleotides GU, and the 3′ end of the intron always bears the nucleotides AG. In the acceptor region, there are two other conserved elements upstream of the AG dinucleotide: the branchpoint sequence (BPS), which lies 18 to 40 bases upstream the AG, and the polypyrimidine (Py) tract placed downstream the BPS. The splicing process begins with a cut at the donor splice site of the pre-mRNA. The excised left end of the intron becomes linked by a 5′–2′ bond to an adenosine of the BPS (branch site) forming a lariat. Then, a second cutting at the 3′ splice site releases the lariat intron, and the downstream exon is joined to the upstream exon. Other sequence motifs involved in exon definition and activation of splice site selection by stimulation of spliceosome assembly are known as exonic splicing enhancers (ESE), as they usually lie within exons. Conversely, sequences that act by blocking the spliceosome assembly are called exonic splicing silencers (ESS). If a single nucleotide substitution or an insertion/deletion of a few nucleotides alters one of the previously mentioned conserved splice site sequences as well as an ESE or ESS element, this will produce aberrant splicing of the pre-mRNA, leading to deleterious effects on protein product. For example, mutations that alter either the AG or the GU splice sites always lead to complete absence of normal splicing (see Fig. 1 ). Indeed, the spliceosome is no more able to recognize the exon and may neglect it, leading to exon skipping, or may use potential splice sites that are not normally selected, termed cryptic splice sites. These can be present in other positions within either the same exon or the flanking introns. Exon skipping and the use of cryptic splice sites are harmful, because they alter the normal ORF of the mRNA by adding pieces of intronic sequences or deleting either the entire exon or a segment of it. The resulting mutant mRNA can be: out-of-frame, thus leading to PTC formation, which is predicted to result in the translation of a truncated unstable polypeptide or NMD activation, or in-frame, and thereby compatible with the synthesis of a protein with internal amino acid deletion or insertion. When a mutation affects the splice site consensus sequence in positions other than the strictly conserved AG/GU dinucleotides, or disrupts the BPS, which is not as well-conserved in people as in yeast, its effects can be leaky or compensated by an alternative element in the vicinity, thus producing only moderately reduced splicing efficiency. The presence of mutant in-frame transcripts or the maintenance of a minimal rate of normal splicing is compatible with the translation of a certain amount of either normal or partially functional protein products. Reduced amounts of a given protein or no protein at all often make the difference between severe and mild/moderate phenotypes of various EB subtypes, as in the case of laminin chain subunits in Herlitz JEB versus non-Herlitz JEB or type VII collagen in severe generalized RDEB versus milder generalized or localized RDEB variants.
Another manner through which a point mutation can affect splicing is the creation of new splice sites. These mutations are single nucleotide substitutions occurring in introns, usually in the vicinity of canonical splice sites, or within exons. When located in an exon, they often may appear as nonpathogenic sequence variants, because they do not change the amino acid (silent mutation), or may be wrongly classified as nonsense, missense or frameshift mutations. Thus, the consequences of a point mutation on splicing should be verified by amplification of the cDNA (the complementary copy of the mRNA) whenever a source of mRNA, such as a skin biopsy or cultured fibroblasts and keratinocytes, is available. This is strongly recommended in particular when a mutation in an ESE or the creation of a new splice site by an apparently harmless nucleotide substitution is suspected. Computational tools for the analysis of the information content and effect prediction of splice site sequence changes are now available, however. Information content splice site analysis can be freely performed via the Web interface at www.fruitfly.org/seq_tools/splice.html or www.cbs.dtu.dk/services/NetPGene/ for splice site prediction, and http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi for ESE prediction.
Splicing mutations are believed to account up to 50% of all mutations in a given gene, and most of those identified in EB patients behave in a recessive manner. However, a few splicing mutations that affect the triple-helix domain of type VII collagen and result in in-frame deletion of X-Y-Gly repeats have a dominant pattern of inheritance and cause DDEB.
Transcription Mutations
All point mutation types described previously fall in the coding region and result in a mutant protein with altered function. Point mutations, however, also may occur 5′ to the start codon or in residues at the 3′ end, in DNA sequences that are critical for regulating transcription. The consequence of these mutations is a decrease or an increase of the amount of a given protein, leading to a loss of normal function (common in recessive diseases) or, more rarely, gain of new biologic function (typical in some autosomal dominant traits), respectively. A single regulatory mutation in the 5′ promoter region that abolishes transcription of a COL7A1 allele has been shown to cause RDEB.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


