Abstract
The cutaneous mucinoses are a heterogeneous group of disorders characterized by an abnormal accumulation of mucin within the skin. While the precise pathogenesis is unknown, cytokines (e.g. TNF, interleukin-1, transforming growth factor (TGF)-β) and/or polyclonal and monoclonal immunoglobulins may induce the synthesis of glycoaminoglycans. The cutaneous mucinoses are divided into: (1) primary mucinoses, in which the mucin deposition leads to clinically distinctive lesions and is the major histologic feature; and (2) secondary mucinoses, in which the mucin deposition is simply an associated finding. Primary mucinoses are further divided into degenerative–inflammatory forms (dermal or follicular) and hamartomatous–neoplastic forms. Major associated disorders include paraproteinemia (scleromyxedema, scleredema), diabetes mellitus (scleredema), thyroid disease (pretibial myxedema, generalized myxedema), and autoimmune connective tissue disease (lupus erythematosus, dermatomyositis).
Keywords
mucin, glycosaminoglycans, dermal mucinoses, follicular mucinoses, scleromyxedema, papular mucinosis, acral persistent mucinosis, self-healing cutaneous mucinosis, lichen myxedematosus, reticular erythematous mucinosis, dysthyroidotic mucinoses, scleredema, pretibial myxedema, myxedema
- ▪
The cutaneous mucinoses are a heterogeneous group of disorders in which an abnormal amount of mucin accumulates in the skin
- ▪
The etiopathogenesis of cutaneous mucinoses is unknown
- ▪
The cutaneous mucinoses are divided into two groups: (1) primary cutaneous mucinoses, in which the mucin deposition leads to clinically distinctive lesions and is the major histologic feature; and (2) secondary mucinoses, in which the mucin deposition is simply an associated finding
- ▪
Primary cutaneous mucinoses are further divided into degenerative–inflammatory forms (which may be dermal or follicular) and hamartomatous–neoplastic forms
- ▪
Associated disorders include paraproteinemia (scleromyxedema, scleredema), diabetes mellitus (scleredema), thyroid disease (pretibial myxedema, myxedema) and autoimmune connective tissue disease (lupus erythematosus, dermatomyositis)
Introduction
The cutaneous mucinoses are a heterogeneous group of disorders in which an abnormal amount of mucin accumulates in the skin, either diffusely or focally .
Mucin is a component of the dermal extracellular matrix and is normally produced in small amounts by fibroblasts. It is a jelly-like, amorphous mixture of acid glycosaminoglycans (formerly referred to as acid mucopolysaccharides), which are complex carbohydrates composed of multiple repeating polysaccharide units (see Ch. 95 ). The acid glycosaminoglycans may be attached to both sides of a protein core (proteoglycan monomer), as in the case of dermatan sulfate and chondroitin sulfate, or they may be free, as in the case of hyaluronic acid, which is the most important component of dermal mucin.
Mucin is capable of absorbing 1000 times its own weight in water, playing a major role in maintaining the salt and water balance of the dermis. In routinely stained sections, the presence of either a blue-staining material between separated collagen bundles or empty spaces within the dermis are good clues that there is mucin deposition. For confirmation, special stains can be used, such as Alcian blue, colloidal iron or toluidine blue ( Table 46.1 ). Furthermore, dermal mucin is PAS-negative and, if composed of hyaluronic acid, hyaluronidase-sensitive. Fixation of biopsy specimens in absolute alcohol, rather than formalin, may improve dermal mucin detection. In addition, monoclonal antibodies have been used to detect sulfated acid glycosaminoglycans . An alternative approach is to utilize biotinylated hyaluronan (HA)-binding proteins coupled to an avidin–peroxidase reaction .
| STAINING CHARACTERISTICS OF ACID GLYCOSAMINOGLYCANS (MUCOPOLYSACCHARIDES) | ||
|---|---|---|
| Histologic stain | Acid glycosaminoglycans (mucopolysaccharides) | |
| Non-sulfated (hyaluronic acid * ) | Sulfated (heparan sulfate, dermatan sulfate, chondroitin sulfate † ) | |
| Colloidal iron | ⊕ | ⊕ |
| Alcian blue | ||
| pH 2.5 | ⊕ | ⊕ |
| pH 0.5 | ⊖ | ⊕ |
| Metachromasia with toluidine blue | ||
| pH 4.0 | ⊕ | ⊕ |
| pH <2.0 | ⊖ | ⊕ |
| PAS ‡ | ⊖ | ⊖ |
| Hyaluronidase-sensitive | ⊕ | ⊖ |
* Major GAG in the disorders discussed in this chapter; not bound to protein core.
† Major GAGs in the mucopolysaccharidoses, e.g., Hunter syndrome, Hurler syndrome; bound to protein core (proteoglycan).
Why mucin abnormally accumulates within the skin of some individuals is unclear. Certain “serum factors”, e.g. immunoglobulins and/or cytokines, could promote the upregulation of glycosaminoglycan synthesis . For example, elevated serum immunoglobulin levels (monoclonal or polyclonal) and circulating autoantibodies can be found in association with cutaneous mucinoses such as scleromyxedema, Graves disease-associated pretibial myxedema, and papulonodular mucinosis of lupus erythematosus. However, the serum from these patients stimulates mucin production even after the elution of either the IgG paraprotein in those with scleromyxedema or the autoantibodies in those with Graves disease-associated pretibial myxedema. Circulating cytokines such as interleukin (IL)-1, TNF-α and TGF-β, which are known to stimulate glycosaminoglycan synthesis within the skin, could play a role in excess mucin deposition. A reduction in the normal catabolic degradation of mucin could also be a factor.
This chapter focuses on the disorders characterized by abnormal dermal deposits of mucin, composed primarily of hyaluronic acid ( Fig. 46.1 ). The mucopolysaccharidoses, in which the predominant dermal mucin is dermatan sulfate or heparan sulfate (e.g. Hunter syndrome), are discussed in Chapter 48 .
Classification
The cutaneous mucinoses may be classified as primary , in which mucin deposition is the major histologic feature resulting in clinically distinctive lesions ( Table 46.2 ), or secondary , in which mucin simply represents an associated histologic finding ( Table 46.3 ). Primary mucinoses can then be divided into degenerative–inflammatory forms and hamartomatous–neoplastic types. The former are further subdivided into dermal and follicular forms, based upon the location of the mucin (see Table 46.2 ).
| CLASSIFICATION OF THE PRIMARY CUTANEOUS MUCINOSES |
| Degenerative–inflammatory mucinoses |
| Dermal |
|
| Follicular |
|
| Hamartomatous–neoplastic mucinoses |
|
* Has been referred to as diffuse/generalized and sclerodermoid lichen myxedematosus.
| DISORDERS ASSOCIATED WITH HISTOLOGIC DEPOSITION OF MUCIN (SECONDARY MUCINOSES) |
| Epithelial mucinosis |
|
| Dermal mucinosis |
|
| Follicular mucinosis |
|
Primary Degenerative–Inflammatory Mucinoses
Dermal Mucinoses
Scleromyxedema
▪ Papular mucinosis ▪ Diffuse/generalized and sclerodermoid lichen myxedematosus ▪ Arndt–Gottron syndrome
Introduction and definition
Scleromyxedema is a chronic idiopathic disorder characterized by numerous firm papules and areas of induration that are due to dermal mucin deposition in association with an increase in dermal collagen . Nearly all patients also have a monoclonal gammopathy, and some may have systemic, even lethal, manifestations . This entity must be distinguished from localized variants of lichen myxedematosus where the skin is the sole site of involvement ( Table 46.4 ). There are, however, occasional patients who have an atypical constellation of findings and fall between scleromyxedema and localized lichen myxedematosus (see “atypical forms” below).
| DIAGNOSTIC CRITERIA OF SCLEROMYXEDEMA VERSUS LOCALIZED VARIANTS OF LICHEN MYXEDEMATOSUS | |
|---|---|
| Scleromyxedema | Localized variants of lichen myxedematosus |
| Generalized papular eruption and sclerodermoid features | Papular eruption (or nodules and/or plaques due to confluence of papules) |
| Microscopic triad (mucin deposition, fibroblast proliferation, fibrosis) or occasionally, interstitial granulomatous pattern | Mucin deposition with variable fibroblast proliferation |
| Monoclonal gammopathy | Absence of monoclonal gammopathy |
| Absence of thyroid disorder | Absence of thyroid disorder |
History
Although the first descriptions of scleromyxedema were attributed to Dubreuilh in 1906 and Reitman in 1908, it was not until the review of Montgomery and Underwood in 1953 that the disease was distinguished from scleroderma and generalized myxedema. A year later, Gottron and his colleagues gave the name “scleromyxedema” to the generalized and sclerotic form. The association of a monoclonal gammopathy with scleromyxedema was first described in 1963.
Epidemiology and pathogenesis
Scleromyxedema is a fairly uncommon disease, affecting middle-aged adults of both sexes equally. The pathogenesis of scleromyxedema is unknown. The role of the associated monoclonal gammopathy remains a matter of debate. While most patients have a plasma cell dyscrasia, paraprotein levels do not correlate with either the extent or the progression of the disease. In addition, sera from patients with scleromyxedema enhanced fibroblast proliferation in vitro , but an immunoglobulin purified from paraprotein-containing sera failed to do so, suggesting a pathogenetic role for circulating factors other than the paraprotein. Clinical remission of scleromyxedema, along with a reduction in the M-protein, that follows autologous hematopoietic stem cell transplantation (HSCT) points to the bone marrow as a source of these circulating factors. Lastly, the development of scleromyxedema following a cutaneous granulomatous reaction to intradermal injections of hyaluronic gel or after silicone breast implants suggests autoimmune phenomena .
Clinical features
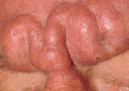
In scleromyxedema, numerous, 2–3 mm, firm, waxy, closely aligned papules develop in a relatively widespread symmetrical pattern. The most common sites of involvement are the head and neck region, upper trunk, hands, forearms and thighs ( Fig. 46.2 ). Papules are often arranged in a strikingly linear array. The surrounding skin is shiny and indurated, i.e. sclerodermoid in appearance, and the glabella is typically involved with deep longitudinal furrowing. Severe involvement of the face can result in a leonine facies ( Fig. 46.3A ). Deep furrowing can also occur on the trunk and extremities and is referred as the “Shar-Pei sign” ( Fig. 46.3B ).


Erythema, edema and a brownish discoloration may also be seen in the involved areas, and pruritus is not uncommon. The mucous membranes and scalp are spared. As the condition progresses, erythematous and infiltrated plaques may appear with skin stiffening, sclerodactyly, and decreased motility of the mouth and joints. Overlying the proximal interphalangeal joints, a central depression surrounded by an elevated rim (due to skin thickening) can be seen and is referred to as the “doughnut sign”. Mat and cuticular telangiectasias and calcinosis, as seen in systemic sclerosis, are absent.
Scleromyxedema is almost always associated with paraproteinemia. The monoclonal gammopathy is usually IgG and the light chains are more commonly lambda. Although a mild plasmacytosis may be observed in bone marrow biopsies, <10% of patients with scleromyxedema progress to symptomatic myeloma. Patients with scleromyxedema can have a number of internal manifestations, in particular muscular, neurologic, rheumatologic, pulmonary, renal and cardiovascular. Dysphagia, proximal muscle weakness due to myositis, disturbances of the CNS leading to unexplained coma, peripheral neuropathy, arthropathies, carpal tunnel syndrome, restrictive or obstructive lung disease, and scleroderma-like renal disease may accompany or follow the cutaneous manifestations . While the predominantly sensory peripheral neuropathy typically affects older men and has an insidious onset, the dermato-neuro syndrome is a potentially life-threatening encephalopathy. This syndrome begins abruptly with a worsening of skin lesions, a flu-like prodrome, fever and seizures, and it can eventuate in an unexplained coma.
Pathology
Scleromyxedema is characterized by a triad of microscopic features :
- •
a diffuse deposit of mucin in the upper and mid reticular dermis
- •
an increase in collagen deposition
- •
a marked proliferation of irregularly arranged fibroblasts ( Fig. 46.4 ).

Fig. 46.4
Scleromyxedema – histopathologic features.
Typical triad of fibrosis, an increased number of irregularly arranged fibroblasts, and interstitial deposits of mucin in the upper and mid reticular dermis.
Courtesy, Lorenzo Cerroni, MD.
The epidermis may be normal or thinned by the pressure of the underlying mucin and fibrosis; the hair follicles may be atrophic. A slight superficial, perivascular, lymphoplasmacytic infiltrate is often present. The elastic fibers are fragmented and decreased in number. More recently, an interstitial granuloma annulare-like pattern was described . Mucin may fill the walls of myocardial blood vessels as well as the parenchyma of the kidney, pancreas, adrenal glands, nerves, and lymph nodes. In the dermato-neuro syndrome, autopsy findings have not proved helpful in elucidating its underlying pathogenesis.
Differential diagnosis
The primary differential diagnosis for scleromyxedema is systemic sclerosis (scleroderma) and scleredema. The presence of papules, especially in linear arrays, is a very helpful clinical sign in distinguishing scleromyxedema. Additional entities in the sclerodermoid differential diagnosis should be excluded (see Ch. 43 ). For example, nephrogenic systemic fibrosis, which develops in individuals with renal impairment exposed to gadolinium-containing contrast media, may show mucin in biopsy specimens, but patients lack both facial involvement (commonly seen in scleromyxedema) and paraproteinemia. Criteria for diagnosing scleromyxedema versus localized variants of lichen myxedematosus are summarized in Table 46.4 , and the dermatologic disorders in which leonine facies can develop are outlined in Table 46.5 .
Treatment
Recommendations are still based on case reports and open-label small case series. In the past, monthly courses of melphalan were often the therapy of choice, targeting the plasma cell dyscrasia. However, while this alkylating agent can result in some clinical improvement, it has also been implicated in 30% of the deaths secondary to its induction of hematologic malignancies and septic complications . Other chemotherapeutic agents (e.g. cyclophosphamide, methotrexate, chlorambucil, 2-chlorodeoxyadenosine) have been tried, but with no better results and the risk of significant side effects.
IVIg, alone or in combination with systemic medications (see below), has gained widespread acceptance as first-line therapy for both cutaneous involvement and associated systemic manifestations, including the dermato-neuro syndrome . Although long-lasting remissions after cessation of IVIg therapy have been reported, the response is usually not permanent and maintenance infusions are required . Thalidomide (or lenalidomide) and/or systemic corticosteroids, standard therapies for multiple myeloma, are considered second-line treatments and are often administered in combination with IVIg . Autologous HSCT represents third-line therapy, especially for individuals with disabling or potentially life-threatening disease . Post-transplant recurrences can be treated with bortezomib plus dexamethasone .
Additional therapies, including PUVA, UVA1, systemic retinoids, cyclosporine, electron beam radiation, plasmapheresis, and extracorporeal photochemotherapy, have all produced variable results. Dysarthria and a flu-like illness may herald the life-threatening coma, and the patient should be promptly admitted to the hospital for close observation. Occasionally, spontaneous improvement and clinical resolution, even after 15 years, have been described .
Lichen myxedematosus (localized variants)
Introduction
In the localized variants of lichen myxedematosus, patients develop small, firm, waxy papules (or nodules and plaques produced by the confluence of papules) that are limited to only a few sites – usually the upper and lower extremities and/or trunk. The skin is the only site of involvement and these variants, in contrast to scleromyxedema, are not associated with sclerosis, paraproteinemia or systemic involvement nor are they associated with thyroid disease (see Fig. 46.1 ) . While most dermatologists equate lichen myxedematosus with localized, skin-limited disease, a source of potential confusion is the uncommon and more historical use of the term “diffuse/generalized and sclerodermoid lichen myxedematosus” to describe scleromyxedema.
The localized variants of lichen myxedematosus are subdivided into four subtypes:
- •
a discrete papular form
- •
acral persistent papular mucinosis
- •
cutaneous mucinosis of infancy
- •
a pure nodular form.
Localized variants of lichen myxedematosus may be observed in association with HIV infection, exposure to toxic oil or L-tryptophan (historical), and hepatitis C viral (HCV) infection.
Epidemiology
Exact incidence and prevalence rates for the localized variants of lichen myxedematosus are not known.
Clinical features
Discrete papular lichen myxedematosus is characterized by 2–5 mm papules, numbering from just a few to hundreds and involving the limbs and trunk in a symmetrical pattern ( Fig. 46.5 ). The affected skin is not indurated and the face is spared. The lesions progress slowly without systemic involvement. However, they rarely resolve spontaneously. To date, progression to scleromyxedema has never been proven.










