Abstract
The term “mosaic” refers to individuals composed of cells with different genotypes that are derived from a genetically homogeneous zygote. Mosaicism can be functional (epigenetic) as in lyonization as well as genetic. Mosaic skin conditions may follow the lines of Blaschko, which are thought to represent migration pathways of epidermal cells during embryogenesis, or have distribution patterns that correspond to body segments or dermatomes. Cutaneous and extracutaneous manifestations of mosaic conditions depend on the timing of onset and the cell type(s) involved. This chapter reviews mosaicism in X-linked dominant and autosomal dominant genodermatoses, including type 1, type 2, and revertant mosaicism. Postzygotic mutations that activate the RAS/mitogen-activated protein kinase (MAPK) and AKT/phosphatidylinositol-3-kinase (PI3K) signaling pathways represent major causes of epidermal nevi with or without associated systemic findings. The postzygotic mutations underlying vascular malformations, pigmented lesions, and benign tumors with mosaic distribution patterns are outlined. Mosaic presentations of inflammatory dermatoses and pigmentary mosaicism are also discussed.
Keywords
mosaicism, Blaschko’s lines, lines of Blaschko, lyonization, functional mosaicism, somatic mutation, postzygotic mutation, type 1 mosaicism, type 2 mosaicism, loss of heterozygosity, revertant mosaicism, incontinentia pigmenti, Goltz syndrome, epidermal nevus, nevus sebaceus, nevus comedonicus, phakomatosis pigmentokeratotica, Proteus syndrome, pigmentary mosaicism, mosaic RASopathies
- ▪
Mosaicism for genes expressed in the skin presents as a patchy distribution of affected and unaffected skin
- ▪
The classical pattern follows the lines of Blaschko but varies according to cell type and timing of mosaicism
- ▪
Epidermal and vascular nevi represent mosaicism for a variety of genes with corresponding associated features
- ▪
Some linear disorders are recognizable as localized forms of autosomal dominant skin disorders, including polygenic inflammatory conditions
- ▪
If there is also gonadal mosaicism, offspring may have the corresponding generalized condition
Introduction and History
The term “mosaic” is used to describe individuals composed of cells of different genotypes that are derived from a genetically homogeneous zygote. For example, patients with mosaic Turner syndrome have both 45,XO and 46,XX cells. The skin provides an ideal canvas to visualize patterns of mosaicism.
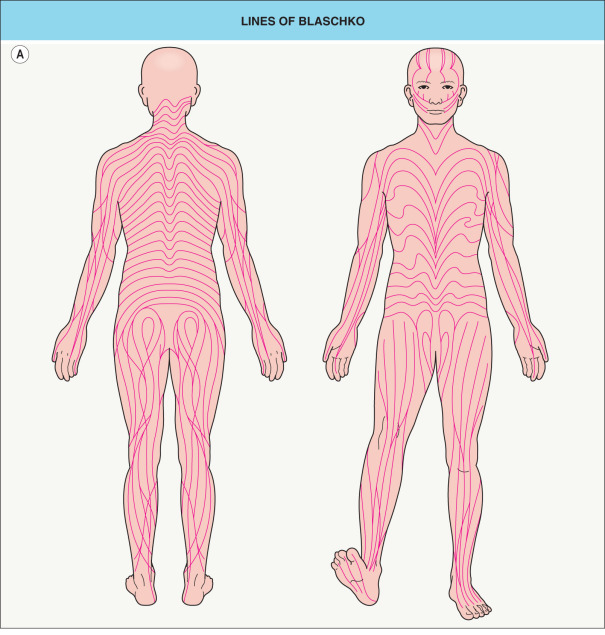
In 1901, Alfred Blaschko documented the characteristic lines and whorls that are followed by epidermal nevi and other skin conditions now known to result from mosaicism ( Fig. 62.1A ). In 1961, Mary Lyon postulated that similar striped patterns in female mice heterozygous for coat color genes on the X chromosome reflected two populations of cells, one expressing the maternal X chromosome and one the paternal X chromosome. She hypothesized that all women are functional mosaics with regard to the X chromosome. In 1965, Curth and Warburton used the Lyon hypothesis to explain the linear pattern of skin findings in the X-linked disorder incontinentia pigmenti. A decade later, Happle recognized lyonization as the reason that skin lesions follow the lines of Blaschko in heterozygous female patients with other X-linked genodermatoses .
We now recognize that mosaicism affecting autosomes as well as X chromosomes may result from either: (1) changes in the DNA sequence due to mutations or chromosomal alterations ( genetic or genomic mosaicism); or (2) changes in gene expression ( epigenetic or functional mosaicism) as in lyonization ( Table 62.1 ). Autosomal mosaic conditions usually result from de novo postzygotic mutations and are not inherited. However, the clinical manifestations of some autosomal dominant disorders reflect a mosaic “second hit” in affected tissues, such as the skin lesions of neurofibromatosis type 1 (NF1; see type 2 mosaicism below). The unusual familial occurrences of autosomal mosaic disorders may be explained by inheritance of either an unstable “premutation” or a transposon (“jumping gene”) of retroviral origin that can modulate the expression of other genes when the disorder has a genetic versus epigenetic etiology, respectively .
| CAUSES OF MOSAICISM AND CHIMERISM | |||
|---|---|---|---|
| Cause | Timing | Pathogenesis | Consequence |
| Genetic (genomic) | |||
| Half-chromatid mutation | First meiotic division of gametogenesis | An incorrect base is added, resulting in a mismatched double strand. When this separates in mitosis, it provides two templates that are not exactly complementary, giving rise to two lines of daughter cells | An individual composed of two different cell lines in approximately equal proportions |
| Somatic mutation | Postzygotic (i.e. any time after fertilization) | Any DNA change | A mutant clone within an otherwise normal individual |
| Chromosomal non-disjunction | Postzygotic | Chromosomes fail to separate correctly during either meiosis or mitosis, resulting in daughter cells with aberrations in chromosome number or structure | An individual composed of two or more different cell lines |
| Chimerism | Pre- or post-zygotic | Fertilization of one egg by two sperm, or fusion of two zygotes | An individual composed of two cell lines with completely different genotypes |
| Epigenetic (functional) | |||
| Lyonization | Early embryonic development | Random inactivation in each cell of either the paternal or maternal X chromosome, which then forms the Barr body; once this occurs, it is the same in all daughter cells | A female composed of 2 different cell lines, expressing either the maternal or paternal X chromosome |
| Epigenetic modification | Inherited or postzygotic | A switch in gene expression, usually involving DNA packaging, resulting from DNA methylation or histone modifications; passed on to daughter cells | Cells with gene expression different from the other cells in that individual |
The idea that the lines of Blaschko represent the embryonic migration pathways of skin cells was first expressed in English by Douglass Montgomery, who proposed that the linear patterns of epidermal nevi “may be due to the streams or trend of growth of the tissues” . Although both Montgomery and Blaschko presented their findings in May 1901, Montgomery’s contribution was eclipsed by that of his German contemporary. In fact, the now widely held theory that the lines of Blaschko reflect routes of embryonic cell migration did not surface again until the late 1970s .
Patterns and Pathogenesis of Cutaneous Mosaicism
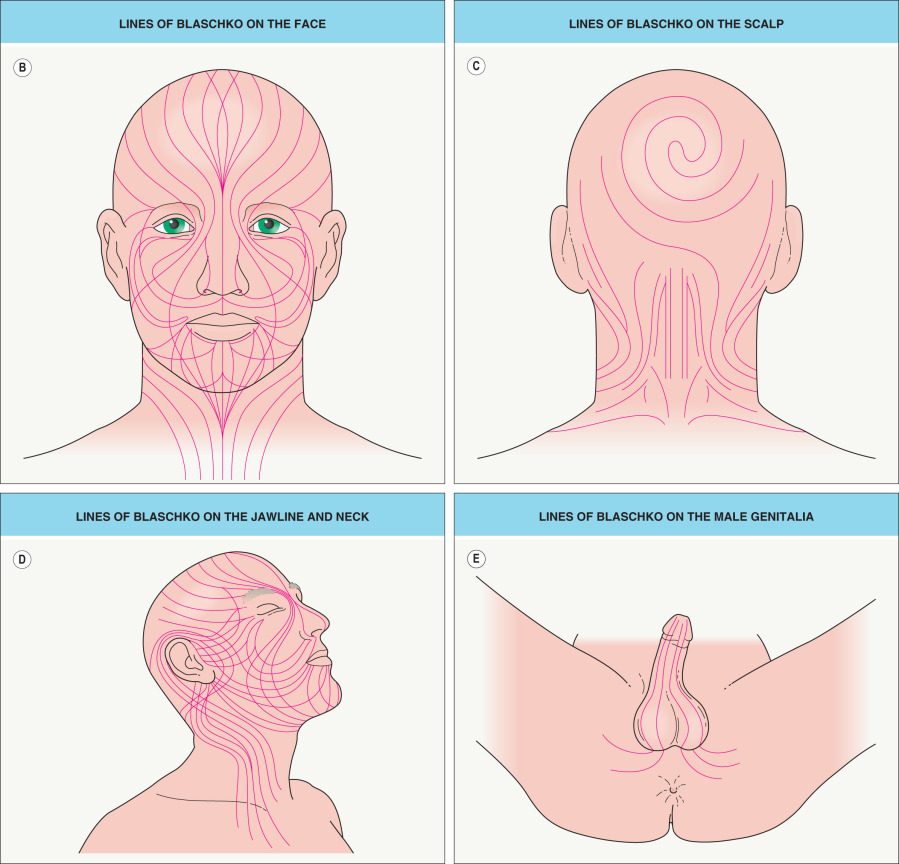
Blaschko’s original diagram (see Fig. 62.1A ) omitted the midface, scalp, lateral neck, and genital area. The trajectories of the lines of Blaschko in these areas have now been established ( Fig. 62.1B–E ), as have analogous patterns in the eyes, teeth, and oral mucosa. Considerable variability occurs in the degree of whorling on the flanks and the directions of lines on the face; the posterior midline may be shifted to the left or right, but the anterior midline tends to be sharply centered from the upper sternum to the pubis.
Mosaic skin conditions do not always follow the lines of Blaschko. Assuming that the pattern is determined by cell migration, it depends on the stage of development at which mosaicism arises ( Table 62.2 ) and the cell type affected ( Table 62.3 ). The classic pattern defined by Blaschko is seen in disorders of epidermal cells , which is not surprising since Blaschko based his diagrams on epidermal nevi. Embryonic keratinocytes move outwards from the neural crest by directional proliferation, in more-or-less continuous lines that are deflected by the complex interplay between cell migration and surface remodeling. In contrast, melanoblasts likely migrate individually to the skin, where they then proliferate antenatally. This as well as other characteristics of the aberrant cells and the timing of the genetic alteration may explain the block-like and phylloid patterns seen in some mosaic pigmentary conditions and the patchy or garment-like distribution of larger congenital melanocytic nevi. Cutaneous blood vessels, fibroblasts, other mesodermal derivatives, and nerve cells take different routes; mosaic disorders involving these tissues typically display patterns that correspond to embryonic segments or dermatomes rather than the lines of Blaschko. The distribution of skin lesions along the lines of Blaschko in Goltz syndrome (focal dermal hypoplasia) and linear morphea are thought to reflect mutated genes that are expressed in the epidermis but lead to changes in the underlying dermis.
| EXPECTED PATTERN OF CUTANEOUS MOSAICISM ACCORDING TO ITS TIMING OF ONSET | ||||
|---|---|---|---|---|
| Timing | Configuration/shape of skin lesions | Number of skin lesions | Laterality | Organs affected |
| Meiosis | Linear | Multiple | Bilateral | Multi-organ |
| Very early blastocyst (includes lyonization) | Linear | Multiple | Bilateral | Multi-organ |
| Later blastocyst | Linear | Fewer | Uni- or bilateral | Multi-organ |
| Later in first trimester, pre-organogenesis | Linear or oval | Fewer | Uni- or bilateral | Multi-organ |
| First trimester, post-organogenesis | Linear or oval | Few or single | Uni- or bilateral | Skin only |
| Second/third trimester | Oval or round | Single | Unilateral | Skin only |
| Postnatal | Round (smaller) | Single | Unilateral | Skin only |
| EXPECTED PATTERN OF CUTANEOUS MOSAICISM ACCORDING TO CELL OR TISSUE TYPE | ||
|---|---|---|
| Cell affected | Embryonic movement | Observed pattern(s) |
| Melanoblast | Single cell migration | Pigmentation: lines of Blaschko, block-like, phylloid Congenital melanocytic nevi: patchy, garment-like |
| Keratinocyte | Directional proliferation following surface forces | Lines of Blaschko |
| Fibroblast, fat cell, other mesodermal cells | Multidirectional proliferation following growth of segment | Segmental |
| Nerve cell | Along future dermatomes | Dermatomal |
| Endothelial and other blood vessel cells | Within segments/future dermatomes | Segmental/dermatomal * |
The phenotypic consequences of mosaicism also vary according to whether the trait is dominant or recessive, as well as whether the mutation is “forward” (pathogenic; gain- or loss-of-function) or revertant (correcting; “backward”/“back”) ( Fig. 62.2 ). In type 1 mosaicism , a postzygotic mutation in one allele of the gene that underlies an autosomal dominant genodermatosis (e.g. epidermolytic ichthyosis) can result in one or more regions of affected skin ( Fig. 62.2A ). The latter may have a linear distribution, e.g. an epidermolytic epidermal nevus.
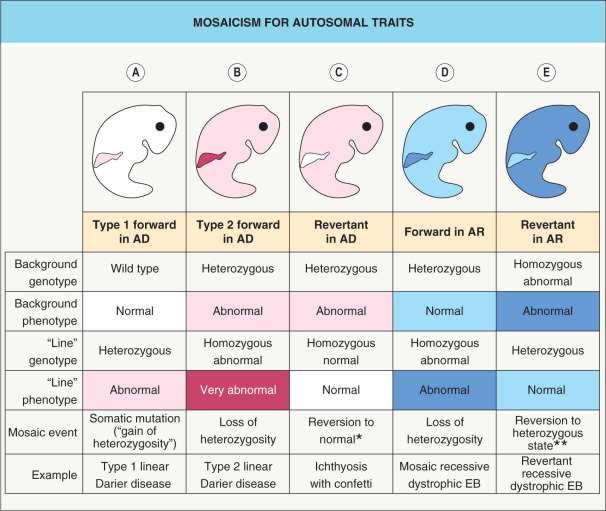
Sometimes, a more severe linear lesion is superimposed upon the background of a generalized autosomal dominant disease. This phenomenon, referred to as type 2 mosaicism , occurs when an individual with a heterozygous germline mutation in the gene for an autosomal dominant disorder develops a postzygotic mutation in the other allele (“second hit”). This leads to a loss of heterozygosity and results in a region (e.g. a linear streak) where the genetic burden is doubled ( Fig. 62.2B ). Type 2 mosaicism has been proven on a molecular level in Hailey–Hailey disease, Darier disease , PTEN hamartoma tumor syndrome , NF1, and basal cell nevus syndrome.
Clonal loss of heterozygosity leading to clinical manifestations may also occur in carriers of autosomal recessive skin conditions such as recessive dystrophic epidermolysis bullosa (EB; Fig. 62.2D ). The opposite phenomenon – regional absence of skin disease attributed to a correcting mutation (revertant mosaicism) – can occur in both autosomal recessive and autosomal dominant conditions, including ichthyosis with confetti and several forms of EB ( Fig. 62.2C, E ). Revertant mosaicism has therapeutic potential and has been employed to create pluripotent stem cells and autologous grafts for patients with recessive dystrophic EB .
Mosaic skin conditions can also be categorized based upon the nature of the corresponding generalized condition ( Table 62.4 ). When assessing a linear skin disorder, it is helpful to consider what the most likely diagnosis would be were the condition to affect the entire skin surface. While the lesional morphology and histology of the mosaic condition are typically similar to the generalized form, the connection may not be immediately obvious due to differences in distribution and overall appearance. If no generalized disorder exists to match the linear lesions, then the patient could have a dominant mutation (autosomal or X-linked) that is only survivable in the mosaic form.
| MOSAIC SKIN CONDITIONS WITH CORRESPONDING GENERALIZED CONDITIONS AND ASSOCIATED GENES | |||
|---|---|---|---|
| Mosaic condition | Generalized condition | Gene symbol [? = likely candidate for mosaic condition] | Protein |
| X-linked dominant disorders: mosaic manifestations in females, usually lethal in males | |||
| Incontinentia pigmenti | (Hypomorphic mutations: hypohidrotic ectodermal dysplasia with immune deficiency) | IKBKG (NEMO) | Inhibitor of κ light polypeptide gene enhancer in B cells, kinase γ (NF-κB essential modulator) |
| Goltz syndrome | NR | PORCN | Porcupine homolog |
| Conradi–Hünermann–Happle syndrome | NR | EBP | Emopamil binding protein |
| CHILD syndrome | NR | NSDHL | NAD(P)-dependent steroid dehydrogenase like |
| MIDAS syndrome | NR | HCCS | Holocytochrome c synthase |
| Oral–facial–digital syndrome type 1 | NR | OFD1 | OFD1 centriole and centriolar satellite protein |
| Terminal osseous dysplasia with pigmentary defects * | NR | FLNA | Filamin A |
| X-linked disorders: variable mosaic manifestations in females, generalized in males | |||
| Linear hypohidrosis, alopecia/lack of adnexa, ± hyperpigmentation | Hypohidrotic ectodermal dysplasia | EDA | Ectodysplasin A |
| Incontinentia pigmenti-like lesions (stages 3 and 4) | Hypohidrotic ectodermal dysplasia with immune deficiency | IKBKG (NEMO) | See above |
| Linear hyperpigmentation | X-linked reticulate pigmentary disorder | POLA1 | DNA polymerase-α1, catalytic subunit |
| Linear hyperpigmentation | X-linked dyskeratosis congenita | DKC1 | Dyskerin |
| Linear hypopigmentation | Menkes disease | ATP7A | ATPase, copper-transporting, α polypeptide |
| Linear hypotrichosis and scaling | Ichthyosis follicularis with atrichia and photophobia (IFAP), X-linked keratosis follicularis spinulosa decalvans, X-linked Olmsted syndrome | MBTPS2 | Membrane-bound transcription factor peptidase, site 2 |
| Autosomal dominant disorders (including lethal conditions) | |||
| Various types of epidermal and sebaceous nevi | See Table 62.7 | ||
| Linear Darier disease | Darier disease | ATP2A2 | ATPase, calcium-transporting |
| Linear Hailey–Hailey disease | Hailey–Hailey disease | ATP2C1 | ATPase, calcium-transporting |
| Linear porokeratosis | Porokeratosis | ? MVK | Mevalonate kinase |
| Linear basal cell nevus | Basal cell nevus syndrome | PTCH1 | Patched 1 |
| Happle–Tinschert/Curry–Jones syndrome | NR | SMO | Smoothened |
| Segmental café-au-lait macules, lentigines, and/or neurofibromas | Neurofibromatosis type 1 | NF1 | Neurofibromin 1 |
| Segmental café-au-lait macules and lentigines | Legius syndrome | SPRED1 | Sprouty-related EVH1 domain-containing 1 |
| Linear/segmental angiofibromas | Tuberous sclerosis complex | ? TSC1, TSC2 | Hamartin, tuberin |
| Segmental leiomyomas | Multiple cutaneous and uterine leiomyomatosis | FH | Fumarate hydratase |
| Linear trichoepitheliomas, spiradenomas, and/or cylindromas | Multiple familial trichoepitheliomas, Brooke–Spiegler syndrome, familial cylindromatosis | ? CYLD | Cylindroma lysine 63 deubiquitinase |
| Venous malformation | Multiple cutaneous and mucosal venous malformations (VMCM), blue rubber bleb nevus syndrome | TEK (TIE2) | TIE-2 endothelial tyrosine kinase |
| Segmental plaque-type glomuvenous malformations | Hereditary glomuvenous malformations | GLMN | Glomulin |
| Verrucous venous malformation (“verrucous hemangioma”) | NR | MAP3K3 | MAPK kinase kinase 3 |
| Parkes Weber syndrome/arteriovenous malformation (AVM) | Capillary malformation (CM)–AVM syndrome | RASA1 | RAS p21 protein activator 1 |
| Sturge–Weber syndrome, port-wine stains, dermal melanocytosis, phakomatosis pigmentovascularis, congenital hemangiomas | NR | GNAQ, GNA11 | Q-type G proteins, α-stimulating |
| Becker melanosis (nevus) | Baraitser-Winter syndrome, juvenile-onset dystonia ** | ACTB | β-actin |
| McCune–Albright syndrome | NR for activating mutations | GNAS | G protein, α-stimulating |
| Classic SLN | Costello syndrome ** | HRAS | HRAS |
| Large/giant CMN, CMN-type SLN | Noonan syndrome ** | NRAS | NRAS |
| Agminated melanocytic nevi; linear syringocystadenoma papilliferum | Cardio-facio-cutaneous syndrome ** | BRAF | BRAF |
| Encephalocraniocutaneous lipomatosis | Craniosynostosis/skeletal dysplasia syndromes ** | FGFR1 | Fibroblast growth factor receptor 1 |
| Multifactorial inflammatory disorder (genetic susceptibility + environmental factors) | |||
| Linear psoriasis Linear lichen planus Segmental vitiligo Linear lichenoid graft-versus-host disease (GVHD) Linear lupus erythematosus and dermatomyositis Linear fixed and lichenoid drug eruptions Linear lichen nitidus Linear morphea Linear atrophoderma of Moulin (similar appearance to atrophoderma of Pasini and Pierini) | |||
* Features hyperpigmented, atrophic (with dermal hypoplasia) facial macules and digital fibromas.
** In general, the mutations in this gene that cause generalized disease are different (e.g. less severe) than those that underlie mosaic disorders.
Mosaicism for autosomal dominant disorders can arise after fertilization (postzygotic/somatic mutation) or less often during gametogenesis (half-chromatid mutation). Although a type 1 mosaic condition itself is not heritable, there is a risk of passing on the full-blown disorder to the next generation if mutant cells are present not just in the skin but also in the gonads (gonadal mosaicism). A correlation between the size or site of the parent’s mosaic lesion and the risk to offspring has not been established. If the parental mutation is known, prenatal diagnosis is possible if clinically appropriate, and men can theoretically undergo DNA analysis of sperm to quantify the risk to their offspring .
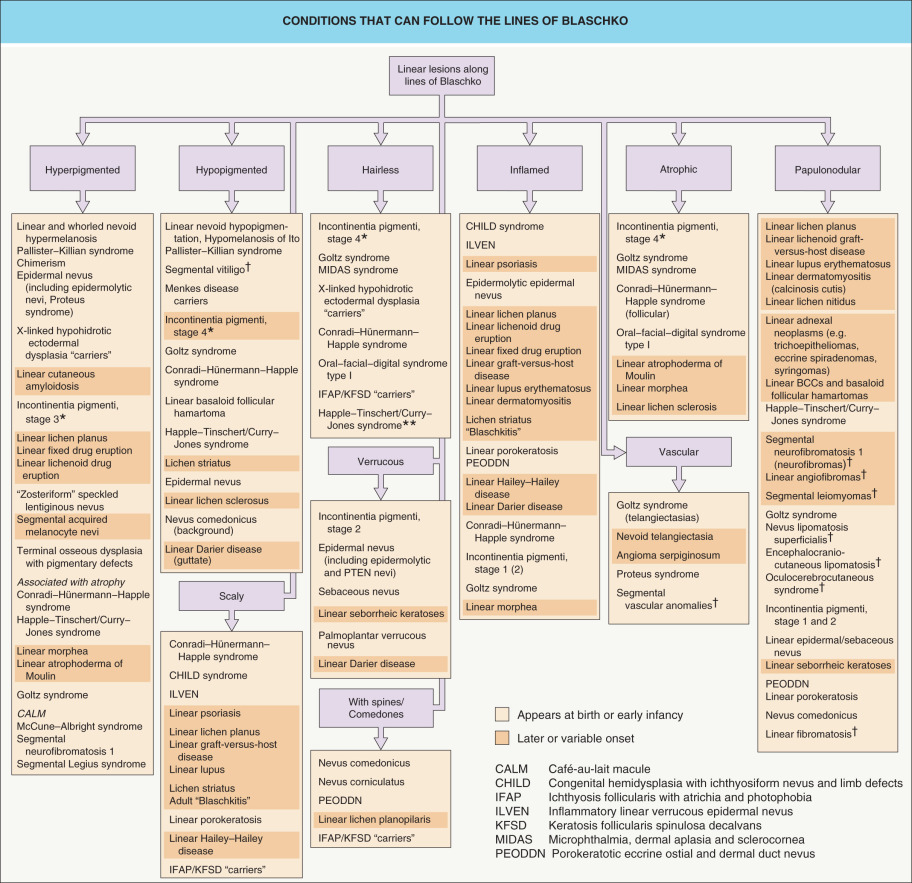
Fig. 62.3 provides an approach to the differential diagnosis of skin lesions that follow the lines of Blaschko. Selected mosaic disorders will be discussed in more detail below.
Mosaicism in X-Linked Disorders
In women heterozygous for X-linked disorders, functional mosaicism due to random X-chromosome inactivation (lyonization) may produce skin lesions following the lines of Blaschko. Sons who inherit the abnormal X chromosome carry and express the mutation in all cells; if “recessive”, the disorder is generally compatible with survival (e.g. hypohidrotic ectodermal dysplasia), but if “dominant”, the disorder is typically so severe that males die before birth (e.g. incontinentia pigmenti, Goltz syndrome). Occasionally, male patients present with skin lesions following the lines of Blaschko because they themselves are mosaic for an X-linked dominant disorder, and therefore, protected by the presence of some normal X chromosomes. X-chromosome mosaicism in boys and men can result from Klinefelter syndrome (where the extra X chromosome permits lyonization) or a postzygotic or half-chromatid mutation (see Tables 62.1 and 62.5 ).
| FINDINGS IN GIRLS AND BOYS WITH NEMO MUTATIONS | |||||
|---|---|---|---|---|---|
| Girl with IP | Boy with IP + Klinefelter syndrome | Son of mother with classic IP | Somatic mosaic boy with IP | Boy with HED-ID † | |
| Karyotype | XX | XXY | XY | XY | XY |
| NEMO genotype | Heterozygous for typical mutation * | Heterozygous for typical mutation * | Hemizygous for typical mutation * | Mosaic hemizygous for typical mutation * (half-chromatid or postzygotic) | Hemizygous for milder (“hypomorphic”) mutation that does not abolish activity |
| Skin findings | Classic IP | Classic IP | NA | Classic IP, although may have a more limited distribution (e.g. unilateral or localized) | Hypotrichosis, hypohidrosis, recurrent pyogenic infections, extensive seborrheic- or atopic-like dermatitis, reticulated hyperpigmentation (less common) |
| Immunodeficiency | No | No | Yes, extremely severe | No | Yes, with dysgammaglobulinemia (↑IgM, ↓IgG) |
| Life expectancy | Normal | Normal | Usually lethal before birth | Normal | Typically shortened due to pyogenic and opportunistic infections; may improve with hematopoietic stem cell transplantation |
* A particular large deletion that removes exons 4–10 is found in ~65% of female patients with IP; a NEMO mutation may be difficult to detect in peripheral blood samples from mosaic male patients (since mutant leukocytes are prone to apoptosis), so analysis of DNA from lesional skin may be required.
† Mother may have a mild variant of IP; affected boys typically have missing/conical teeth. An allelic variant features osteopetrosis and lymphedema, and an autosomal dominant condition with a similar phenotype can result from mutations in NF-κB inhibitor α gene ( NFKBIA ).
Lyonization occurs synchronously in all cells at about the 1000-cell stage, so the two clones are intimately mixed from the beginning. Female patients with X-linked genodermatoses typically have numerous skin lesions that follow the lines of Blaschko in narrow bands. A curious exception is CHILD ( c ongenital h emidysplasia with i chthyosiform nevus and l imb d efects) syndrome, which characteristically shows large unilateral blocks of abnormal skin. This lateralization pattern may reflect the effects of expression of the mutant versus normal X-chromosome in organizer cells that control large developmental fields. Alternatively, a deficiency in sonic hedgehog signaling (which normally helps to confer left–right asymmetry) in cells expressing the NSDHL mutation may result in selection against these cells on one side of the body.
Not all X-linked skin disorders follow the lines of Blaschko. For example, women who are heterozygous for the gene defects that underlie Fabry disease, Wiskott–Aldrich syndrome, and chronic granulomatous disease have either no lesions or scattered lesions in a non-linear pattern. This is likely because the cells that express the mutated gene are circulating leukocytes or non-epidermal cells (e.g. endothelial cells) for which lyonization does not produce a clinically apparent pattern. In the case of steroid sulfatase-deficient ichthyosis, where the defect is epidermal but not evident in female carriers, the steroid sulfatase gene is on a part of the X chromosome that does not undergo lyonization.
Incontinentia Pigmenti
▪ Bloch–Sulzberger syndrome
- ▪
An X-linked dominant disorder with skin lesions that follow the lines of Blaschko
- ▪
Four cutaneous stages: (1) inflammatory/vesicular; (2) verrucous; (3) hyperpigmented; and (4) hypopigmented/atrophic
- ▪
Other ectodermal abnormalities may be present, e.g. alopecia, nail dystrophy, pegged or missing teeth
- ▪
May also have ocular (~20–40%) and neurologic (~30%) involvement
- ▪
Caused by mutations in IKBKG ( NEMO )
Introduction
Incontinentia pigmenti (IP) is a multisystem disorder with an estimated birth prevalence of 1 : 140 000 and a female : male ratio of ~20 : 1. Patients may present to neonatologists, neurologists, ophthalmologists, or dentists as well as dermatologists. However, the diagnosis rests on the cutaneous findings. The name refers to the pathologic finding of pigmentary incontinence, i.e. dermal melanophages, in the third stage of the disease. The linear skin lesions reflect mosaicism secondary to X inactivation (lyonization).
History
Although Garrod probably first described IP in 1906, Bloch’s 1926 report of a “previously unreported pigment disorder” and that 2 years later by Sulzberger are remembered eponymously. In 2000, the International Incontinentia Pigmenti Consortium established that IP is caused by mutations in IKBKG which encodes inhibitor of κ light polypeptide gene enhancer in B cells, kinase γ, also known as NEMO ( N F-κB e ssential mo dulator). A separate condition previously known as “IP1” is now recognized as a form of pigmentary mosaicism (see below).
Genetics
IP is an X-linked dominant disorder, usually antenatally lethal in boys, that is caused by mutations in IKBKG located at Xq28 . Approximately two-thirds of patients have a common large deletion of exons 4–10 that tends to arise during paternal meiosis (male gametogenesis). Occasional occurrences of clinically typical IP in boys are attributed to Klinefelter syndrome (~10% of male patients) or to genomic mosaicism due to a half-chromatid or postzygotic NEMO mutation ( Table 62.5 ) . In the latter patients, the distribution of the skin lesions may be relatively limited.
Pathogenesis
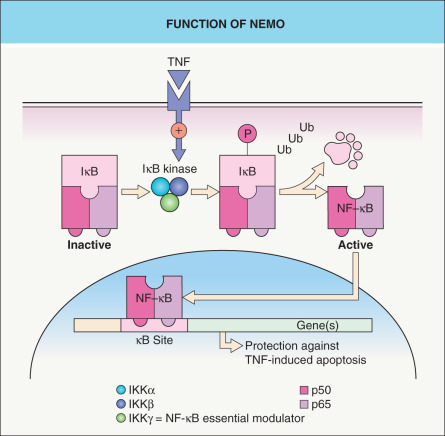
The NEMO protein is a subunit of a kinase that activates NF-κB, a transcription factor that protects against TNF-α-induced apoptosis ( Fig. 62.4 ). The idea of IP as a pro-apoptotic state explains male lethality, destruction of epidermal cells, and progressive replacement of cells expressing the mutant X chromosome by those expressing the normal allele . In one report, a liveborn boy whose mother had classic IP died hours after birth due to lethal hematopoietic and immunologic disturbances . Milder hypomorphic NEMO mutations are responsible for hypohidrotic ectodermal dysplasia with immune deficiency (HED-ID) in boys, whose mothers often have relatively subtle features of IP (see Table 62.5 and Ch. 63 ) .
Clinical features
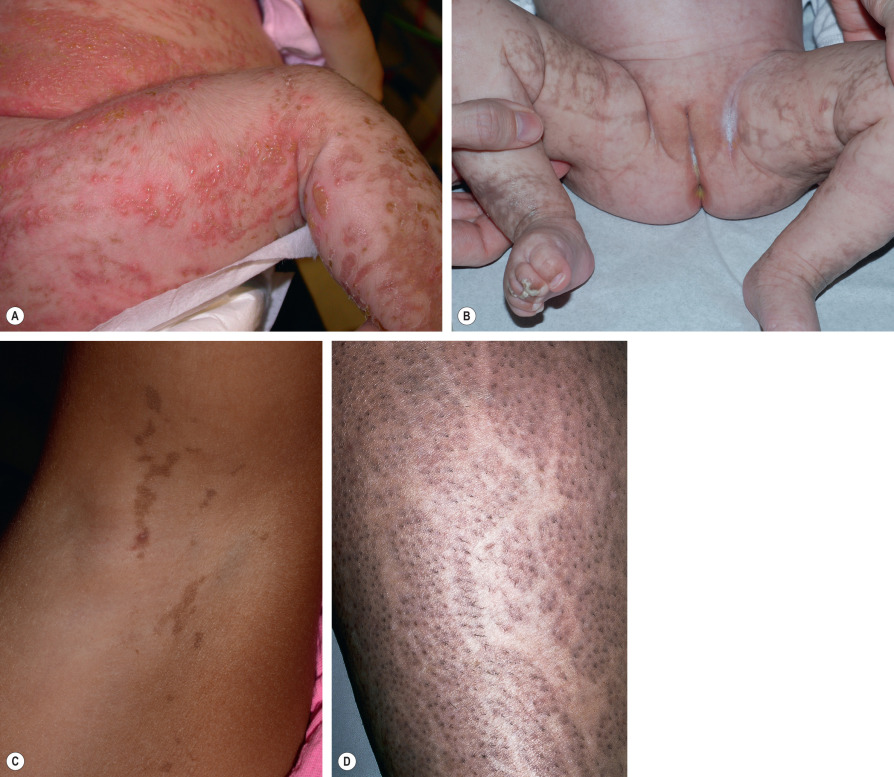
IP typically presents with linear erythema and vesicles (stage 1) during the first few weeks to months of life in an otherwise healthy baby girl (see Fig. 34.13 ) . Vesiculobullous lesions are most common on the limbs and scalp, frequent on the trunk, and rare on the face ( Fig. 62.5A ). They resolve within days to weeks and are often replaced by verrucous linear plaques (stage 2) that favor the extremities ( Fig. 62.5B ) and usually disappear later in infancy. Streaks and swirls of reticulate grayish-brown hyperpigmentation (stage 3) develop next and have a predilection for the trunk and intertriginous sites, with scalloped edges that are thought to reflect the growth of normal keratinocytes into affected skin ( Fig. 62.5B, C ). The inflammatory sequence sometimes recurs within pigmented areas during later infancy or childhood together with intercurrent febrile illnesses. In addition, acral keratotic nodules (including subungual lesions) occasionally arise after puberty. The hyperpigmented streaks tend to fade by adolescence, although a few areas of slate-gray pigmentation may persist lifelong. From puberty onward, linear hypopigmented bands lacking hair and sweat glands (stage 4) appear on the posterior aspects of the limbs (especially the calves; Fig. 62.5D ) and may be the only stigmata of the disease during adulthood . Individual stages of IP may be absent or overlap. Extracutaneous manifestations of this condition are listed in Table 62.6 .
| ADDITIONAL MANIFESTATIONS OF INCONTINENTIA PIGMENTI |
| Surface ectodermal defects |
|
| Eye anomalies (20–40% of patients) |
|
| Central nervous system abnormalities (~30% of patients) |
|
| Skeletal defects (uncommon) |
|
| Other organs (uncommon) |
|
Pathology
The early inflammatory phase of IP demonstrates eosinophilic spongiosis and scattered dyskeratotic keratinocytes. The epidermis of verrucous lesions is acanthotic with hyperkeratosis and foci of dyskeratosis. In stage 3, there is pigmentary incontinence as well as variable vacuolization of basal keratinocytes, whereas stage 4 is characterized by a thinned epidermis and dermis devoid of adnexa .
Differential diagnosis
Neonatal IP can usually be distinguished from infectious conditions (e.g. herpes zoster, varicella, herpes simplex viral infections) by the wellbeing of the child and characteristic patterning of the skin lesions. Peripheral eosinophilia and leukocytosis are common in neonates with IP, and histologic examination can confirm the diagnosis. In linear nevoid hyper- or hypopigmentation, there is no preceding inflammatory phase, the hyperpigmentation is epidermal, and adnexa are typically normal.
Treatment
Baseline and longitudinal ophthalmologic (especially during infancy) and neurologic evaluations are recommended, as are dental assessments and early intervention when anomalies are present. The mother should be examined for subtle atrophic streaks, usually most visible on the calves, and genetic counseling provided. The Incontinentia Pigmenti International Foundation ( www.ipif.org ) is a good resource for families.
Hypohidrotic Ectodermal Dysplasia With Immune Deficiency
This condition is discussed in detail in Chapter 63 .
Goltz Syndrome (Focal Dermal Hypoplasia)
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


