Abstract
The major determinant of normal skin color is the activity of melanocytes, i.e. the quantity and quality of pigment production, not the density of melanocytes. Melanocytes contain a unique intracytoplasmic organelle, the melanosome, which is the site of melanin synthesis and deposition. Compared with lightly pigmented skin, darkly pigmented skin has melanosomes that contain more melanin and are larger; once transferred to keratinocytes, the melanosomes are singly dispersed and degraded more slowly. Tyrosinase is the key enzyme in the melanin biosynthetic pathway and the two major forms of melanin produced in melanocytes are brown–black eumelanin and yellow–red pheomelanin. The production of eumelanin versus pheomelanin is influenced by the binding of melanocyte stimulating hormone to the melanocortin 1 receptor.
Keywords
melanocyte, melanosome, tyrosinase, eumelanin, pheomelanin, melanocortin 1 receptor, MC1R, melanocyte stimulating hormone, MSH, pigmentation, agouti, melanin biosynthetic pathway
- ▪
The major determinant of normal skin color is the activity of melanocytes, i.e. the quantity and quality of pigment production, not the density of melanocytes
- ▪
Melanocytes contain a unique intracytoplasmic organelle, the melanosome, which is the site of melanin synthesis and deposition
- ▪
Compared with lightly pigmented skin, darkly pigmented skin has melanosomes that contain more melanin and are larger; once transferred to keratinocytes, the melanosomes are singly dispersed and degraded more slowly
- ▪
Tyrosinase is the key enzyme in the melanin biosynthetic pathway
- ▪
Two major forms of melanin are produced in melanocytes: brown–black eumelanin and yellow–red pheomelanin
- ▪
The production of eumelanin versus pheomelanin is influenced by the binding of melanocyte stimulating hormone to the melanocortin 1 receptor
Introduction
In order to understand the underlying pathophysiology of cutaneous disorders of hypopigmentation and hyperpigmentation, as well as the process of normal physiologic pigment production, an appreciation of the structure and function of the melanocyte is required. A classic example of basic pathogenesis is type 1 oculocutaneous albinism (OCA1), in which pigmentary dilution of the skin, hair, and eyes is due to a reduction or absence of tyrosinase activity secondary to mutations in both copies of the tyrosinase gene ( TYR ). Within the realm of physiologic pigmentation, melanocytes in individuals with red hair often express variants of the melanocortin 1 receptor (MC1R) . As a consequence of the altered amino acid sequences of the variant MC1Rs, their cell surface expression and interactions with melanocyte stimulating hormone (MSH) can be affected, leading to an increase in the production of pheomelanin as opposed to eumelanin. Based upon population genetics, genes that are mutated in OCA (e.g. TYR , OCA2 , TYRP1 , SLC45A2 , SLC24A5 ) also influence normal pigment variation ( Table 65.1 ) .
| DISORDERS CHARACTERIZED BY DIFFUSE PIGMENTARY DILUTION IN WHICH THE GENETIC DEFECT IS KNOWN | |||
|---|---|---|---|
| Disorder | Gene | Protein product | Comments |
| Oculocutaneous albinism (OCA) [~40% of patients have OCA1 and ~50% have OCA2] | |||
| OCA1A | TYR | Tyrosinase |
|
| OCA1B | TYR | Tyrosinase |
|
| OCA2 | OCA2 | P protein ( OCA2 was previously known as P ) |
|
| OCA3 | TYRP1 | Tyrosinase-related protein 1 * |
|
| OCA4 | SLC45A2 | Solute carrier family 45 member 2 (previously known as MATP) |
|
| OCA5 | Not known | − |
|
| OCA6 | SLC24A5 | Solute carrier family 24 member 5 |
|
| OCA7 | C10orf11 | Chromosome 10 open reading frame 11 |
|
| Hermansky–Pudlak syndrome (HPS) † | |||
| HPS1 | HPS1 | HPS1 (BLOC-3 subunit 1) |
|
| HPS2 | AP3B1 | Adaptor related protein complex 3 β1 subunit |
|
| HPS3 | HPS3 | HPS3 (BLOC-2 subunit 1) |
|
| HPS4 | HPS4 | HPS4 (BLOC-3 subunit 2) |
|
| HPS5 | HPS5 | HPS5 (BLOC-2 subunit 2) |
|
| HPS6 | HPS6 | HPS6 (BLOC-2 subunit 3) |
|
| HPS7 | DTNBP1 | Dystrobrevin binding protein 1 |
|
| HPS8 | BLOC1S3 | BLOC1S3 (BLOC-1 subunit 3) |
|
| HPS9 | BLOC1S6 | BLOC1S6 (BLOC-1 subunit 6) |
|
| HPS10 | AP3D1 | Adaptor related protein complex 3 δ1 subunit |
|
| Chédiak–Higashi syndrome | |||
| LYST | Lysosomal trafficking regulator |
| |
| Griscelli syndrome | |||
| GS1 | MYO5A | Myosin Va (attaches melanosomes to actin filaments) |
|
| GS2 | RAB27A | RAB27A (melanosomal membrane GTPase that binds melanophilin) |
|
| GS3 | MLPH | Melanophilin (links myosin Va and RAB27A) |
|
| MYO5A | Myosin Va F-exon deletion |
| |
* Recognized by Mel-5 antibody.
† Reflecting a founder effect, individuals of Puerto Rican origin have HPS1 and HPS3 (3 : 1 ratio); non-Puerto Ricans most commonly have HPS1, followed by HPS3 and HPS4 with ~70% of patients having one of these three types.
The major sections in this chapter are:
- •
the origin and function of the melanocyte
- •
the formation and function of the melanosome
- •
regulation of melanin biosynthesis.
Origin and Function of the Melanocyte
The melanocyte is a neural crest-derived cell, and during embryogenesis precursor cells (melanoblasts) migrate along a dorsolateral then ventral pathway via the mesenchyme to reach the epidermis and hair follicles of the trunk (see Ch. 2 ). More recently, it was shown that cutaneous melanocytes can also arise from neural crest-derived Schwann cell precursors that migrate along nerves to the skin via a distinct ventral pathway . Additional sites of melanocyte migration include the uveal tract of the eye (choroid, ciliary body, and iris), the leptomeninges, and the inner ear (cochlea) ( Fig. 65.1 ). Presumably, the death of melanocytes within the leptomeninges, inner ear, and skin is responsible for the aseptic meningitis, auditory symptoms, and areas of vitiligo, respectively, seen in patients with the Vogt–Koyanagi–Harada syndrome (see Ch. 66 ).
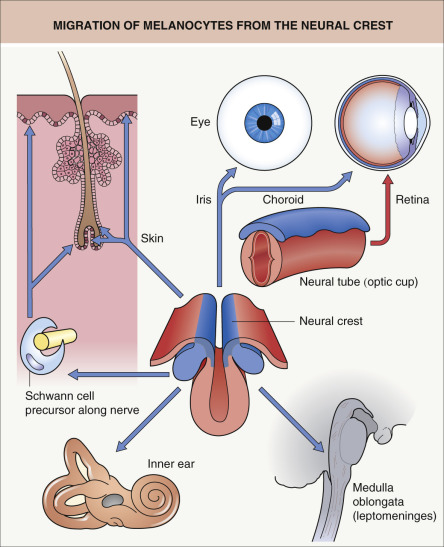
In the inner ear, particularly in the stria vascularis, melanocytes are thought to play a role in the development of hearing. Aberrant migration or survival of melanocytes within the inner ear, the iris, and midportions of the forehead and extremities explains the presence of congenital deafness, heterochromia irides, and patches of leukoderma, respectively, in patients with Waardenburg syndrome, the classic neurocristopathy. Also, aberrant migration or survival of enteric ganglion cells, another neural crest-derived cell population, provides an explanation for the association of aganglionic megacolon (Hirschsprung disease) with Waardenburg syndrome or rarely piebaldism.
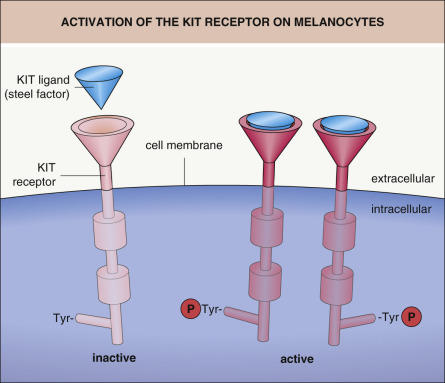
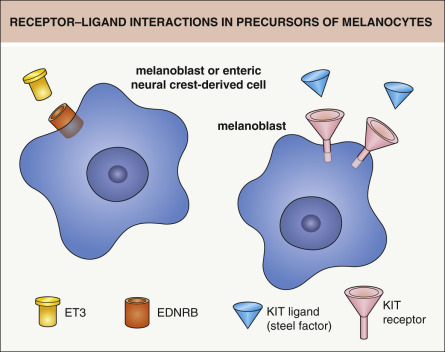
The survival and migration of neural crest-derived cells during embryogenesis depends upon interactions between specific receptors on their cell surface and extracellular ligands. For example, KIT ligand (also known as steel factor or stem cell growth factor) binds to the transmembrane KIT receptor on melanocytes and melanocyte precursors (melanoblasts) ( Figs 65.2 & 65.3 ); melanoblasts require expression of the KIT receptor in order to maintain their normal chemotactic migration directed by production of KIT ligand by the dermamyotome. Heterozygous germline mutations in KIT that decrease the ability of the KIT receptor to be activated by KIT ligand are responsible for human piebaldism, whereas in mice, mutations in either kit or steel can lead to white spotting. In the developing embryo, melanoblasts expressing endothelin receptor type B (EDNRB) are stimulated to migrate by endothelin-3 (ET3 [EDN3]), which is produced by the ectoderm and dermamyotome. Mutations in one or both copies of EDN3 or EDNRB can result in Waardenburg syndrome plus aganglionic megacolon (see Fig. 65.3 ).
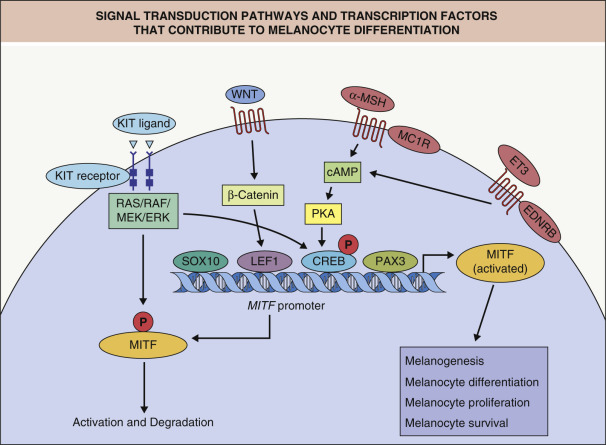
Transcription factors represent another group of proteins that play an essential role during embryogenesis. Because transcription factors can bind DNA and influence the activity of other genes, they are able to regulate the complex interplay of various sets of genes that is required for embryonic development. Several of the genes that, when mutated, give rise to Waardenburg syndrome encode transcription factors (e.g. PAX3, MITF, SOX10 ; see Table 66.4 ). Fig. 65.4 demonstrates some of these interactions (e.g. PAX3 and SOX10 can control expression of MITF) . MITF is sometimes referred to as the master regulator of melanocyte development and function given its modulation of multiple differentiation genes and its early up-regulation in neural crest cells that will eventually become melanocytes and emigrate from the dorsal neural tube.
During embryogenesis, melanin-producing melanocytes are found diffusely throughout the dermis. They first appear in the head and neck region at ~10 weeks of gestation. However, by the end of gestation, active dermal melanocytes have “disappeared”, except in three primary anatomic locations – the head and neck, the dorsal aspects of the distal extremities, and the presacral area . Some of the dermal melanocytes have clearly migrated into the epidermis, but, given the absolute numbers of cells in the two compartments, apoptosis of pigment cells has also occurred. The three sites where active dermal melanocytes are still present at the time of birth coincide with the most common sites for dermal melanocytoses and dermal melanocytomas (blue nevi). Hepatocyte growth factor may play a role in the survival and proliferation of these dermal melanocytes as well as somatic activating mutations in GNA11 and GNAQ , which encode G proteins and are found in blue nevi (see Table 112.3 ).
As depicted in Fig. 65.1 , melanocytes also migrate to the basal layer of the hair matrix and the outer root sheath of hair follicles. Cells that are actively producing melanin are easily recognized in the matrices of pigmented anagen hairs, whereas melanocytes within the outer root sheath are usually amelanotic and more difficult to identify . It has been hypothesized that there are two populations of melanocytes in the skin, one in the interfollicular epidermis and the second in the hair follicle . Based upon antigen expression and clinical observations, it is the former that is more sensitive to the destructive forces of vitiligo. As a result, repigmentation of patches of vitiligo in which the hairs are still pigmented relies upon activation and subsequent upward migration of the melanocytes present in the outer root sheath . Of note, melanocyte stem cells have been identified within the lower portion of the hair follicle bulge, i.e. the lowermost permanent portion of the hair follicle (see Fig. 2.6 ). In addition, KROX20+ cells, which give rise to the hair shaft, have recently been identified within the hair bulb. These hair progenitor cells produce stem cell factor (SCF) which was shown to be essential for hair pigmentation, i.e., mouse hairs turn white when the SCF gene was deleted .
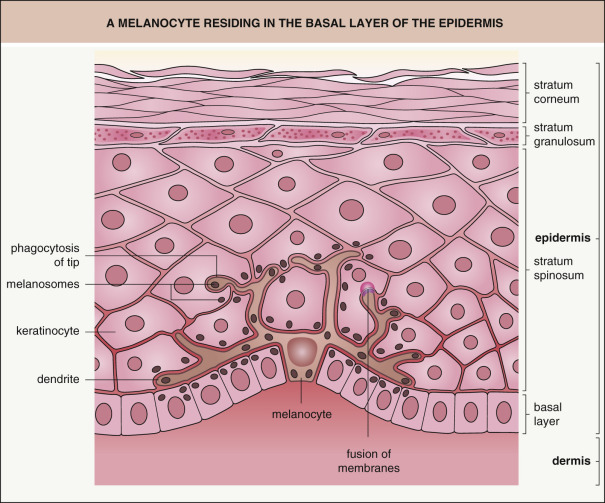
By immunohistochemical staining, melanocytes are identified within the fetal epidermis as early as 50 days of gestation . Melanin-containing melanosomes are recognizable by electron microscopy during the fourth month of gestation. Except in benign and malignant neoplasms, melanocytes reside within the basal layer of the epidermis, a location they maintain throughout life ( Fig. 65.5 ). Although the cell body of the melanocyte sits on a specialized region of basal lamina, its dendrites come into contact with keratinocytes as far away as the mid stratum spinosum. This association of a melanocyte with ~30–40 surrounding keratinocytes to which it transfers melanosomes has been called the epidermal melanin unit . However, melanocytes fail to form desmosomal connections with neighboring keratinocytes; their interactions with keratinocytes are via cadherins.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


