Many medical conditions present with scar-like lesions.
These scar-like lesions often have different pathogenic pathways than scars induced by trauma.
Understanding the similarities and differences in the pathogenic pathways of scar formation and scar-like lesion formation may help us to treat, rehabilitate, and prevent scars and scar-like lesions.
and the dermis and subcutaneous fat are replaced by thick, pale sclerotic collagen bundles (Fig. 3-3). Dermal appendages and blood vessels are replaced by collagen.
Table 3-1 Scar-like conditions organized by pathogenic mechanism | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
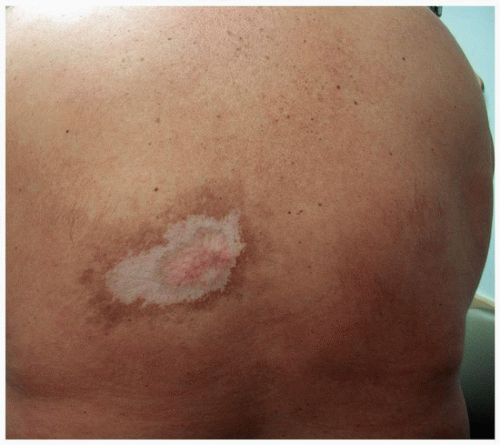 FIGURE 3-1 Morphea—white sclerotic plaque with surrounding hyperpigmentation. |
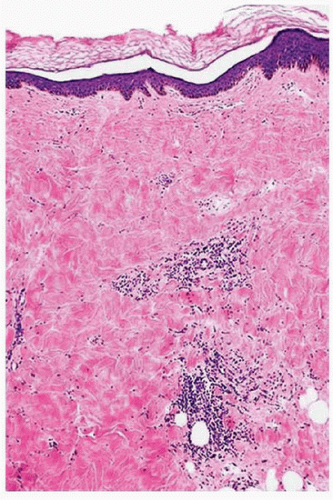 FIGURE 3-2 Morphea early histology—perivascular lymphocytic infiltrate with increased collagen deposition and loss of fat and adnexal structures. |
DNA antibodies, antihistone antibodies, rheumatoid factor, and anti-topoisomerase IIα antibodies.3 The sclerosis in morphea lesions is thought to be initiated by vascular injury via environmental exposure or autoantibodies. Endothelial injury causes release of cytokines that cause increased expression of vascular cell adhesion molecule 1 (VCAM-1), intercellular adhesion molecule 1 (ICAM-1), and E-selectin.3 The result is an initial inflammatory response characterized by TH1+ cells, interleukin (IL)-2, interferon gamma (IFN-γ), and tumor necrosis factor alpha (TNF-α).7 Profibrotic cytokines such as IL-4 and -6 and TGF-β are expressed, which recruit eosinophils, CD4+ T cells, and macrophages.3 Expression of IL-6 is thought to transition the TH1+ environment to a TH17 environment, and then ultimately a TH2+ environment characterized by IL-4 and -13.7 This prosclerotic environment results in increases in collagen, fibronectin, and proteoglycan and a decrease in proteases.
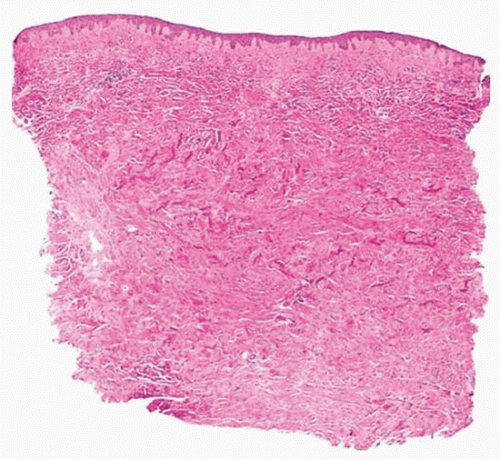 FIGURE 3-3 Morphea late histology—increased dermal collagen with loss of blood vessels, adnexa, and fat. |
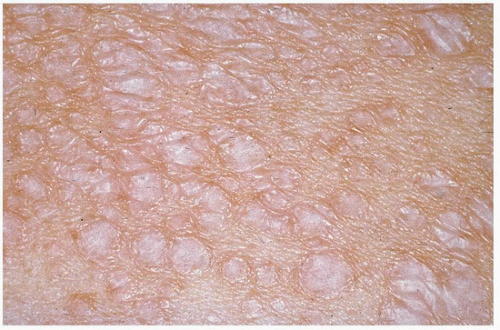 FIGURE 3-4 Lichen sclerosus et atrophicus (LSA)—white thin sclerotic macules with epidermal atrophy. |
anti-RNA polymerase III antibodies (Table 3-2). Patients with LcSScl are more likely to develop isolated pulmonary artery hypertension (which is sclerosis of the pulmonary artery), whereas patients with DcSScl are more likely to develop renal crisis, likely due to sclerosis of the arcuate and intralobular arteries in the glomeruli.25 The organ sclerosis that occurs in systemic sclerosis results in high morbidity and mortality.
Table 3-2 Autoantibodies and Associations | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
and (3) the residual phase—during which collagen formation predominates.33
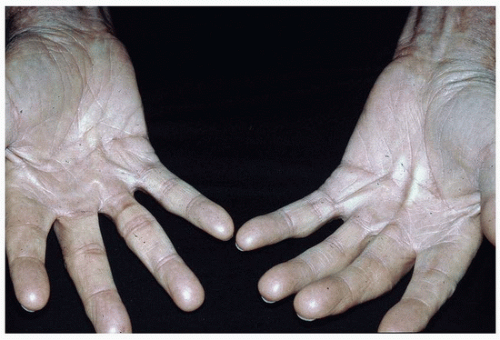 FIGURE 3-5 Dupuytren’s contractures—subcutaneous palmar cords with associated finger contractions. |
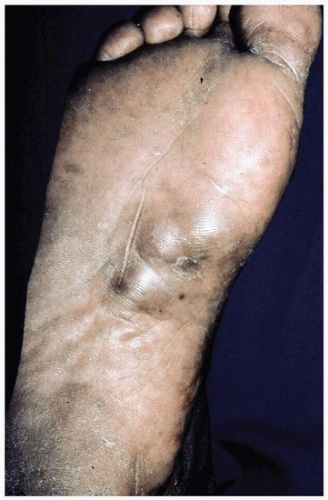 FIGURE 3-6 Plantar fibromatosis—subcutaneous plantar nodules. |
between the ages of 15 and 30 years (Fig. 3-7).42 Knuckle pads have been observed to occur concomitantly with other superficial fibromatoses, although their pathogenesis has not been studied. Knuckle pads are a cutaneous finding of Bart-Pumphrey syndrome (knuckle pads, leukonychia, palmoplantar keratoderma, and deafness caused by an autosomal dominant mutation in the GJB2 gene). Histologically, the nodules are made up of fibroblasts and collagen. Due to their asymptomatic nature and difficult-to-treat location, knuckle pads are not usually treated.
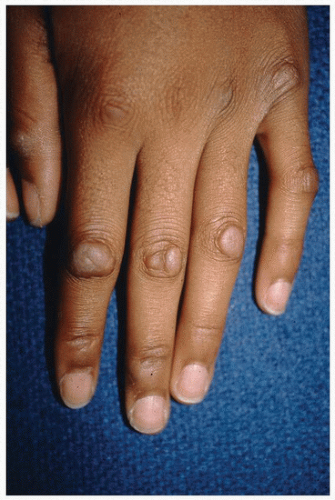 FIGURE 3-7 Knuckle pads—flesh-colored nodules over the extensor hand joints. |
and tends to be refractory to treatment.49 Histopathological findings are the same in all three subsets and include a normal epidermis, a normal number of fibroblasts, and a dermis that is thickened due to an increase in collagen bundles and deposition of GAGs. The pathophysiology of scleredema is not understood and has been incompletely studied. It is postulated that in scleredema secondary to diabetes there is nonenzymatic glycosylation of dermal collagen, which potentially activates fibroblasts resulting in increased collagen production and increased GAGs.50 It is hypothesized that the scleredema that occurs in the setting of a monoclonal gammopathy is due to paraprotein stimulation of fibroblasts.
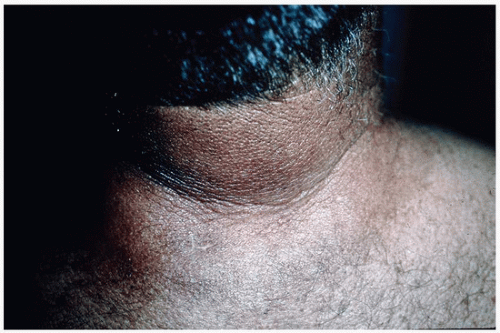 FIGURE 3-8 Scleredema—firm, thickened skin on the posterior neck and upper back. |
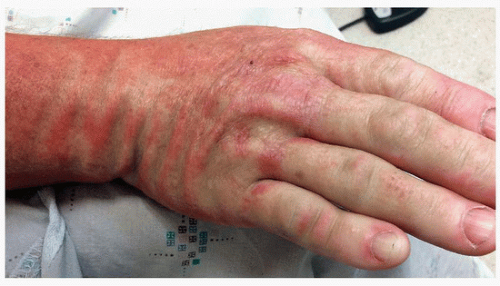 FIGURE 3-9 Scleromyxedema—erythematous edematous plaques. Bending of the skin results in increased furrowing, known as the Shar-Pei sign. |
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


