Abstract
Mastocytosis may occur at any age and results from mast cell proliferation, which can involve the skin, bone marrow, and other organs. Activating mutations in KIT have a pathogenic role in most patients. Childhood disease is most common and frequently resolves by adolescence, with presentations ranging from a solitary mastocytoma to multiple papules or plaques (“urticaria pigmentosa”) to diffuse cutaneous involvement. Adult disease persists throughout life and has variable systemic and cutaneous involvement, with the latter typically appearing as multiple small, red–brown macules or papules. Treatment is focused on controlling symptoms related to mast cell mediator release with antihistamines and other modalities.
Keywords
mastocytosis, mast cell disease, mastocytoma, urticaria pigmentosa, telangiectasia macularis eruptiva perstans, Darier’s sign, tryptase, KIT receptor, tryptase, indolent systemic mastocytosis, smoldering systemic mastocytosis, well-differentiated systemic mastocytosis
- ▪
Mastocytosis can develop from birth to adulthood and may involve only the skin (most children and some adults) or multiple organs such as the bone marrow, liver, spleen, and/or lymph nodes (primarily adults)
- ▪
Childhood disease is more common and often presents with one or more tan to brown papules or plaques (“urticaria pigmentosa”) that frequently resolve by adolescence
- ▪
Mastocytomas are thicker plaques or nodules that occur primarily in children
- ▪
Adults with mastocytosis may or may not have cutaneous lesions; when present, they typically appear as small red–brown macules or papules. Adult disease persists throughout life
- ▪
Stroking of lesions of mastocytosis often causes urtication (Darier’s sign), which is more pronounced in children due to a higher density of mast cells
- ▪
Activating mutations in codon 816 of KIT are detected in the majority of adults and up to 40% of children with mastocytosis; extracellular domain mutations also occur frequently in childhood-onset disease
- ▪
Patients may have accompanying symptoms of mast cell mediator release such as pruritus, flushing, headaches, abdominal pain, diarrhea, and syncope
- ▪
Treatment is primarily directed at controlling symptoms
Introduction
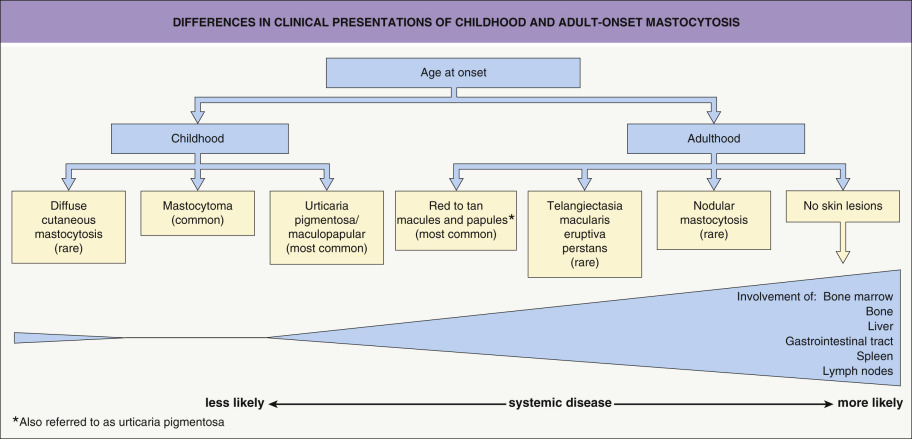
Mastocytosis or mast cell disease represents a spectrum of clinical disorders with a common phenotype of tissue mast cell hyperplasia that can involve the skin, bone marrow, and other organs. Despite significant advances in understanding the pathogenesis of mastocytosis, explanations for the differences in clinical presentations and course between childhood-onset and adult-onset disease are still needed ( Fig. 118.1 ). Childhood mastocytosis has an excellent prognosis, with ~50–70% of affected children experiencing resolution by adolescence. In contrast, adult-onset mastocytosis typically has a chronic course and is more often associated with extracutaneous involvement and systemic symptoms . Therapy for mastocytosis currently centers on inhibiting the effects of secreted mast cell mediators , with a subgroup of patients responding temporarily to tyrosine kinase inhibitors such as imatinib. Cytoreductive therapies (e.g. cladribine) are reserved for advanced systemic disease .
History
The original description of mastocytosis was by Nettleship and Tay, who told of a 2-year-old girl with hyperpigmented papules that spontaneously urticated . Eight years later, in 1877, Paul Ehrlich formally discovered the mast cell. The next year, Sangster suggested the name “urticaria pigmentosa” . Unna was the first to demonstrate that mast cells were responsible for the cutaneous eruption in mastocytosis patients.
Epidemiology
Mastocytosis can be present at birth or develop any time thereafter, including into late adulthood. Childhood-onset mastocytosis is defined as presenting prior to puberty. In a pediatric series (n=101), cutaneous mastocytosis appeared by ages 6 months and 2 years in 73% and 97% of children, respectively . Adult-onset mastocytosis most often develops in the third or fourth decade of life . Mastocytosis has no gender preference, and it occurs in all races .
While most individuals with mastocytosis have no family history of the disorder, there have been >70 familial cases reported, including at least 15 sets of monozygotic twins; however, twin pairs discordant for the disease have also been described. Mastocytosis has been documented in three generations of a single kindred .
Pathogenesis
Mast cells are derived from pluripotent CD34 + precursors in the bone marrow that then circulate in the peripheral blood as agranular, monocytic-appearing cells. After migrating into tissues, these immature mast cells assume their typical granular morphology (see Ch. 18 ). Circulating mast cell precursors express CD34, the tyrosine kinase receptor KIT (CD117), and IgG receptors (FcγRII), but not high-affinity IgE receptors (FcεRI) . The KIT receptor is expressed on mast cells, melanocytes, primitive hematopoietic stem cells, primordial germ cells, and interstitial cells of Cajal. Activation of KIT by its ligand (stem cell factor [SCF]) induces mast cell growth and maturation, and it extends cell survival by preventing apoptosis. Membrane-bound and soluble forms of SCF exist, and both induce KIT activation . SCF is produced by bone marrow stromal cells, fibroblasts, keratinocytes, endothelial cells, and reproductive Sertoli and granulosa cells. Peripheral blood mast cell precursors cultured in the presence of SCF and other cytokines (e.g. interleukin [IL]-3, -4, -6, -9) become KIT + /CD34 − /FcγRII − /FcεRI + and develop characteristic cytoplasmic mast cell granules, as seen in mature tissue mast cells .
Alterations in KIT structure and activity are central to the pathogenesis of mastocytosis. Somatic activating mutations in KIT involving codon 816 represent the most common genetic abnormality in patients with sporadic mastocytosis. The result is a substitution of the amino acid aspartic acid (D) with valine (V, i.e. D816V) or another amino acid ( Fig. 118.2 ). These mutations lead to constitutive, ligand-independent activation of the receptor. Other less common activating mutations and a rare dominant inactivating mutation have also been identified in children and adults with mastocytosis (see Fig. 118.2 ) . In addition, rare germline mutations have been reported .
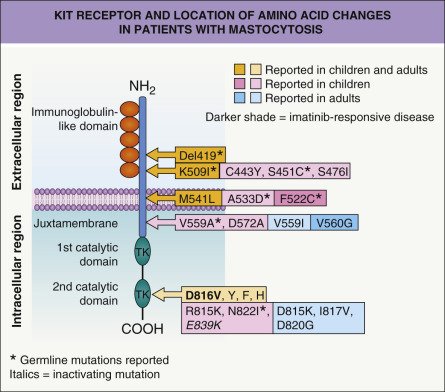
In a study in which the entire KIT sequence was analyzed in skin biopsy specimens from 50 children (ages 0–16 years) with cutaneous mastocytosis (mostly “urticaria pigmentosa”), codon 816 mutations were found in 42% and other activating mutations in 44% of the patients . These results obviously led to a shift in thinking about the basis for spontaneously resolving childhood-onset mastocytosis.
In a transgenic mouse model of mastocytosis in which human activated KIT (D816V) was expressed, only 8 of 28 genetically identical animals developed clinical features of mastocytosis, ranging from indolent mast cell hyperplasia to an invasive mast cell tumor . This diverse phenotypic expression, along with the lack of KIT mutations in some patients and families with mastocytosis, points to additional pathogenic factors. Indeed, in adult patients with advanced systemic mastocytosis, somatic mutations have been identified in both KIT and a variety of other genes such as TET2, ASXL1, JAK2, SRSF2, RUNX1, and CBL , which encode Tet methylcytosine dioxygenase 2 , a dditional s e x combs l ike 1 , transcriptional regulator, Ja nus k inase 2 , s erine and arginine r ich s plicing f actor 2 , runt related transcription factor 1, and Cbl proto-oncogene, respectively .
Clinical Features
Signs and Symptoms
Many children and adults have few, if any, symptoms. When symptoms do occur, they are due to the diverse physiologic effects of secreted mast cell mediators, such as histamine, eicosanoids, and cytokines ( Fig. 118.3 ). These complaints and findings may range from pruritus and flushing to abdominal pain and diarrhea to palpitations, dizziness, and syncope. Of interest is the relative absence of pulmonary symptoms in mastocytosis. Complaints of fever, night sweats, malaise, weight loss, bone pain, epigastric distress, and problems with mentation (cognitive disorganization) often signal the presence of extracutaneous disease. There are even rare reports of deaths in children and adults in association with extensive mast cell mediator release.
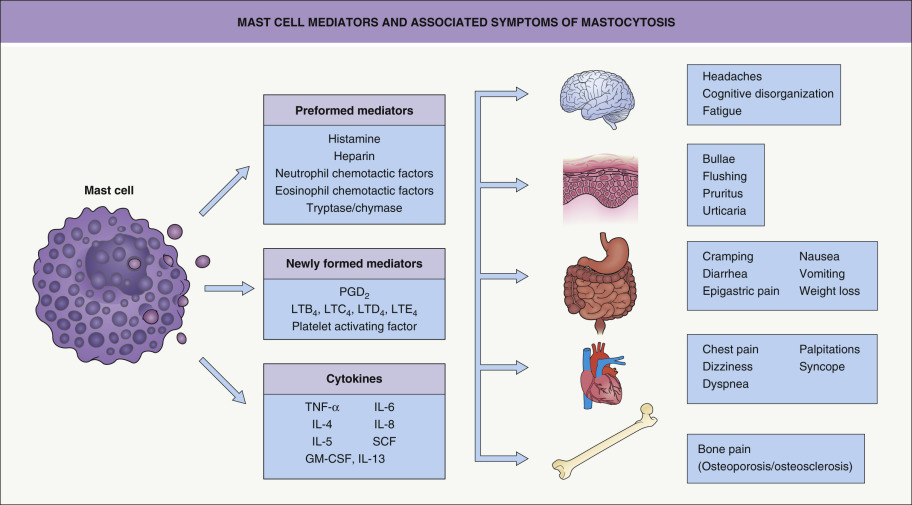
Symptoms of mastocytosis can be exacerbated by exercise, heat, or local trauma to skin lesions. In addition, alcohol, narcotics, salicylates and other nonsteroidal anti-inflammatory drugs (NSAIDs), polymyxin B, and anticholinergic medications have been implicated in precipitating symptoms of mastocytosis. Some systemic anesthetic agents may induce anaphylaxis (see below).
Cutaneous Lesions
Childhood-onset mastocytosis, which almost always affects the skin, has three main clinical presentations: (1) solitary or few (≤3) lesions, referred to as “mastocytomas” – 15–50% of patients ; (2) multiple (<10 to >100) lesions, referred to as “urticaria pigmentosa” (UP) or “maculopapular” mastocytosis – 45–75% of patients; and (3) diffuse cutaneous involvement – <5–10% of patients . A mastocytoma appears as a tan or yellow–tan to brown nodule or plaque, which may be only slightly elevated and typically has a leathery texture ( Fig. 118.4 ). These lesions can be congenital or become evident during infancy, and they favor the distal extremities .
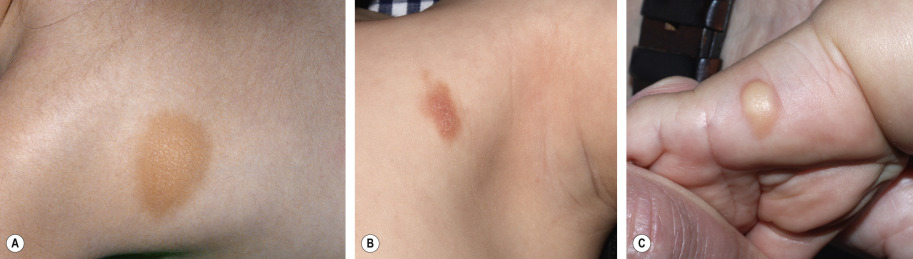
Two major variants have been described in children with multiple lesions of mastocytosis: small, monomorphic tan to brown macules or papules, similar to the lesions typically observed in adults (see below); and larger, polymorphic yellow–tan to brown plaques or nodules, which may be admixed with smaller lesions ( Fig. 118.5 ). Multiple lesions favor the trunk and classically spare the central face, palms, and soles. The large/polymorphic variant is seen predominately in children, and it is associated with an earlier onset (e.g. in infancy) and is more likely to spontaneously resolve .
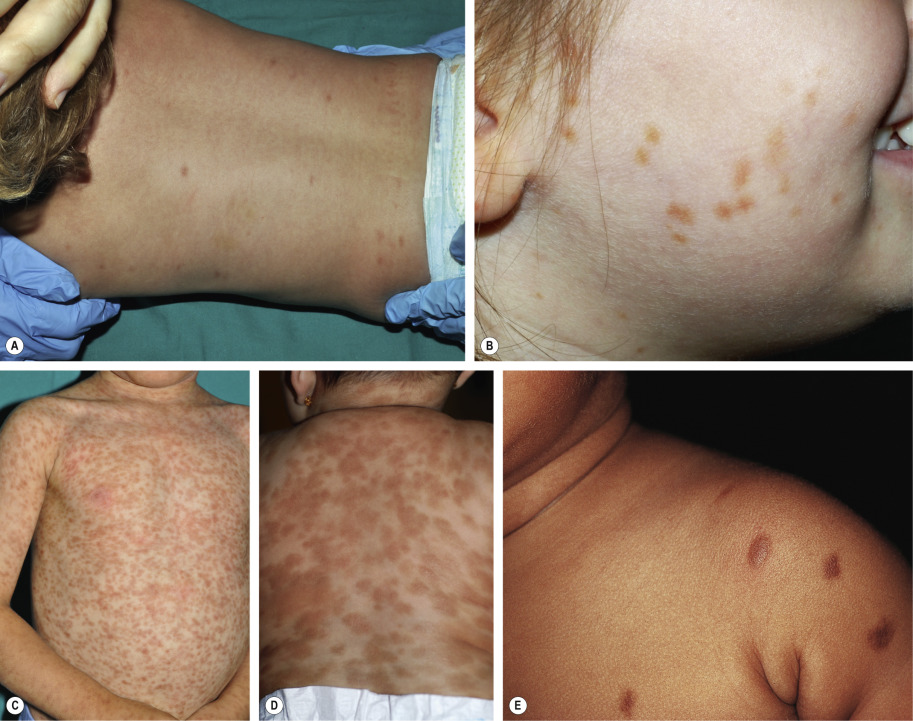
Diffuse cutaneous mastocytosis presents within the first few months of life as thickened skin with a leathery texture and variable hyperpigmentation in a generalized distribution ( Fig. 118.6 ); however, systemic disease is typically absent and spontaneous resolution usually occurs . This term should be reserved for diffuse skin involvement rather than the confluence of individual lesions. Less common presentations of mastocytosis in children include ill-defined tan or telangiectatic patches; small, firm brown nodules; and yellowish patches with superimposed doughy papules (“xanthelasmoid” mastocytosis).
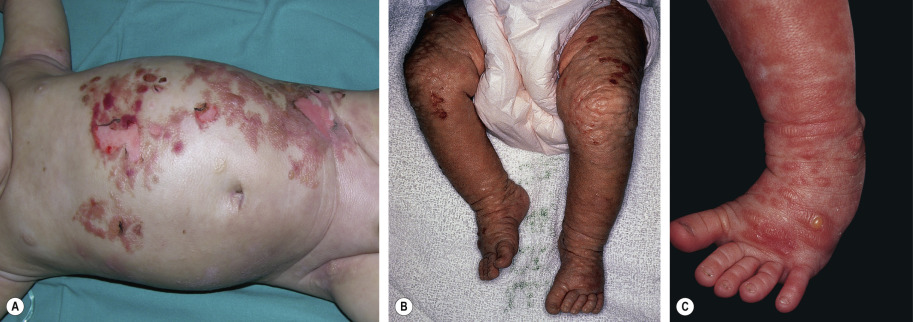
Blistering following urtication is common in infants and young children with mastocytosis, especially those with large, thick lesions or the diffuse form. The latter patients may present with extensive bullae and erosions (see Fig. 118.6 ; see Ch. 34 ). The tendency to blister remits by 3 to 5 years of age, and it is believed to result from the release of mast cell serine proteases.
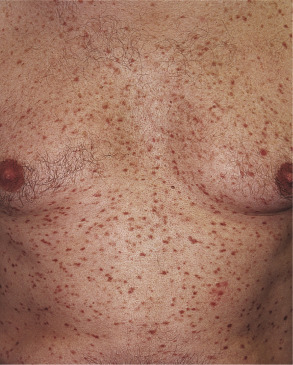
The most common cutaneous lesions of adult-onset mastocytosis are small (<1 cm), monomorphic, reddish-brown macules and papules ( Fig. 118.7 ); in patients with skin type I, they may be pink to red in color. These lesions are most numerous on the trunk and proximal extremities and appear less frequently on the face, distal extremities, or palms and soles. Individual lesions can resolve, but the total number usually increases over time. Close inspection reveals variable hyperpigmentation and fine telangiectasias. These lesions are most often associated with indolent systemic mastocytosis (see below).
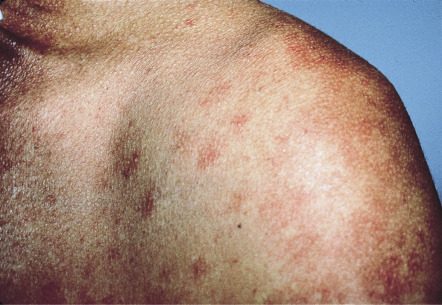
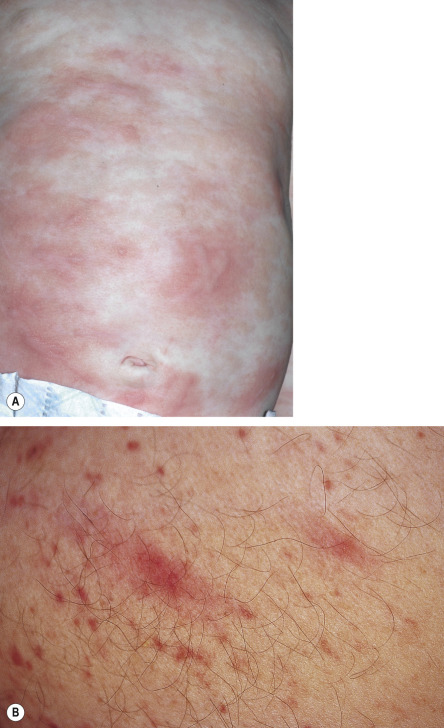
Less commonly, yellow–tan to red–brown nodules or papulonodules develop, representing the hallmark of well-differentiated systemic mastocytosis (see below). This variant has a female predominance, frequently represents continuation of childhood disease, and is less often associated with codon 816 KIT mutations . Severe infiltrative mastocytosis may progress to diffuse thickening of the skin, especially in flexural sites; facial involvement may have a leonine appearance. A rare form of adult cutaneous mastocytosis is telangiectasia macularis eruptiva perstans (TMEP), which is characterized by macules and patches composed of telangiectasias without significant hyperpigmentation ( Fig. 118.8 ). Mastocytomas are extremely rare in adults.
Formation of an urticaria-like wheal upon firmly stroking or rubbing of skin lesions (Darier’s sign) results from release of mast cell mediators and is indicative of the diagnosis ( Fig. 118.9 ). Darier’s sign is readily demonstrated in pediatric patients, especially with nodular lesions, and it may be accompanied by systemic symptoms. It tends to be more subtle in typical adult mastocytosis lesions and barely detectable in TMEP. These differences reflect lesional mast cell density, which is higher in mastocytomas and childhood UP than in adult-onset cutaneous mastocytosis . Dermographism may also be observed in the uninvolved skin of pediatric and occasionally adult mastocytosis patients.
Systemic Manifestations
Skeletal lesions are detected more commonly in adult patients with mastocytosis and virtually never occur in children . In one study of 142 adults with mastocytosis, 40% had skeletal involvement, with a similar frequency in those with childhood-onset and adult-onset disease . Bony lesions may appear as radio-opacities, radiolucencies, or a mixture of the two. The skull, spine, and pelvis are most commonly involved. In an earlier study of 58 adult systemic mastocytosis patients, 57% had diffuse bone involvement, whereas only 2% had focal lesions. Demineralization was the most common change in patients with diffuse skeletal disease, followed by osteosclerosis and mixed lesions of osteosclerosis and osteoporosis . Patients may complain of skeletal pain.
Undecalcified iliac crest biopsies from adult mastocytosis patients have demonstrated increased numbers of mast cells and evidence of enhanced cortical and trabecular bone turnover. These observations led to the hypothesis that mast cells and their mediators are directly responsible for the associated skeletal changes. For example, mast cell-derived heparin and SCF have been implicated in promoting osteoporosis via stimulation of osteoclastic activity. Osteoclasts have been shown to express KIT on their surface and to be activated by SCF. Mast cell histamine, however, is also capable of promoting bone sclerosis through activation of fibroblasts, and mast cell-derived IL-6 appears to induce both bone resorptive and fibrotic activities .
The bone marrow is involved in nearly all adult patients with mastocytosis. In those with indolent systemic disease (form seen in most adult patients with cutaneous mastocytosis), flow cytometry of a bone marrow biopsy specimen is often necessary to detect the associated mild increase in aberrant mast cell number (see below). Some groups recommend performing a bone marrow biopsy in all adult mastocytosis patients . However, others (including the authors) do not believe that a bone marrow biopsy is required in adults with indolent mastocytosis and normal hematologic parameters, especially if there is limited cutaneous disease and serum tryptase levels are normal. In contrast, bone marrow involvement is relatively uncommon in children with cutaneous mastocytosis , and bone marrow biopsy is not routinely recommended since the results do not generally impact disease management or prognosis .
Splenomegaly, detected either clinically or by CT scan, has been reported in 50–60% of adult systemic mastocytosis patients . However, in more recent larger studies, each with >140 adult mastocytosis patients, splenomegaly was observed in only 8–9% of individuals . Increased numbers of mast cells and eosinophils are frequently observed in the spleen, as are various degrees of fibrosis and hematopoiesis. In general, lymph node enlargement is uncommon in mastocytosis patients but it does occur in those with more advanced systemic disease. Among 58 systemic mastocytosis patients, 26% had peripheral lymphadenopathy, whereas 19% had central nodal disease . Histologically, early involvement of lymph nodes often consists of only clusters of mast cells, while in more advanced disease, mast cell infiltrates involve the paracortex and they are often accompanied by eosinophils .
Gastrointestinal (GI) symptoms that are due to mast cell mediator release, such as abdominal pain, diarrhea, nausea and vomiting, may occur spontaneously or may be precipitated by alcohol, aspirin, NSAIDs, or certain foods . Approximately 25% of patients with UP and 70% of those with systemic mastocytosis report GI symptoms . Diarrhea in patients with mastocytosis is usually episodic; it can result from malabsorption, increased motility, and/or acid hypersecretion, with the former due to infiltration and the latter two probably a result of the release of mast cell histamine and prostaglandins. Patients with systemic mastocytosis are at increased risk of gastritis and peptic ulcers, which occasionally lead to GI hemorrhage . A number of radiographic changes in the GI tract have been described, including urticaria-like lesions and thickened gastric, duodenal and jejunal folds, as well as mucosal nodules and/or peptic ulcers. Biopsies of mucosal nodules have demonstrated numerous mast cells with varying numbers of eosinophils. Gastrointestinal stromal tumors (GISTs) frequently have underlying somatic activating KIT mutations and can occur together with cutaneous mastocytosis in families with germline activating KIT mutations (see Fig. 118.2 ) . Hepatomegaly occurs in 10–40% of systemic mastocytosis patients, but abnormal liver function tests are detected less frequently .
A mixed organic brain syndrome with a constellation of symptoms – including irritability, fatigue, headache, poor attention span and motivation, limited short-term memory, inability to work effectively, and difficulty in interacting with other people – has been described in patients with mastocytosis . It has been hypothesized that these symptoms may be secondary to released mast cell mediators. Electroencephalographic studies in these patients range from normal to changes consistent with a toxic or metabolic process.
Classification of Mastocytosis
The World Health Organization (WHO) classification scheme, proposed in 2001 and modified in 2008, includes seven mastocytosis variants ( Table 118.1 ) . Cutaneous mastocytosis represents the largest group and includes nearly all affected children and some adults. Childhood-onset mastocytosis typically has a limited course, following one of three paths: (1) disease resolution by adolescence – ~50–70% of patients; (2) persistence into adulthood – <10–15% of patients; and (3) a marked reduction in lesion number in the remainder . With underlying somatic KIT mutations in the majority of children with mastocytosis , the reasons for its frequent spontaneous resolution remain unknown.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


