Abstract
This chapter describes the clinical features, etiologies, evaluation, and management of ichthyoses and erythrokeratodermas. Both groups represent disorders of cornification in which abnormal differentiation and desquamation of the epidermis results in a defective cutaneous barrier. Ichthyoses are characterized by generalized scaling of the skin with or without an erythematous component, while the hallmark of erythrokeratoderma is circumscribed areas of erythema and hyperkeratosis, usually without substantial scale. Entities that are discussed include the most common disorders, ichthyosis vulgaris due to filaggrin deficiency and X-linked recessive ichthyosis due to steroid sulfatase deficiency; nonsyndromic conditions such as keratinopathic ichthyoses and autosomal recessive congenital ichthyoses (ARCI) due to defective epidermal lipid metabolism and transport; various syndromic ichthyoses; and disorders of cornification due to connexin defects or abnormal cholesterol metabolism. The underlying genetic defects and pathomechanisms are highlighted, as this provides a basis for the development of targeted treatments.
Keywords
ichthyosis, erythrokeratoderma, disorder of cornification, ichthyosis vulgaris, steroid sulfatase deficiency, epidermolytic ichthyosis, ichthyosis with confetti, ichthyosis hystrix Curth–Macklin, ichthyosis hystrix, collodion baby, self-improving collodion ichthyosis, autosomal recessive congenital ichthyosis, lamellar ichthyosis, congenital ichthyosiform erythroderma, Netherton syndrome, Sjögren–Larsson syndrome, neutral lipid storage disease, trichothiodystrophy, erythrokeratodermia variabilis, progressive symmetric erythrokeratoderma, keratitis– ichthyosis–deafness syndrome, CHILD syndrome, Conradi–Hünermann–Happle syndrome, Harlequin ichthyosis
- ▪
Ichthyoses and erythrokeratodermas are heterogeneous groups of disorders of cornification
- ▪
Ichthyoses are characterized by scaling of the skin in a widespread distribution
- ▪
Erythrokeratodermas feature discrete areas of erythema and hyperkeratosis, usually without substantial scale
- ▪
Most inherited ichthyoses and erythrokeratodermas are evident at birth or manifest in infancy
- ▪
Clinical features and pattern of inheritance as well as the underlying structural, biochemical, and molecular abnormalities help to differentiate these disorders
- ▪
Therapy is symptomatic and primarily aimed at reducing hyperkeratosis. Topical management consists of emollients, keratolytics, and retinoids
- ▪
Many, but not all, disorders respond well to oral retinoids. Treatment is initiated at low doses and titrated to response. Benefits and side effects must be carefully considered because of the chronic nature of these conditions
Introduction
Ichthyoses and erythrokeratodermas are disorders of cornification in which abnormal differentiation and desquamation of the epidermis result in a defective cutaneous barrier. Ichthyoses represent a large clinically and etiologically heterogeneous group of conditions that feature generalized scaling of the skin ( Table 57.1 ). Erythrokeratodermas are characterized by circumscribed areas of erythema and hyperkeratosis without obvious scaling. Willan introduced the term “ichthyosis”, derived from the Greek root “ ichthy ” meaning fish, in his textbook of dermatology in 1808 . Since then, nomenclature and nosology of ichthyoses have continuously evolved, including a variety of descriptive names, eponyms, and synonyms.
| FEATURES OF SELECTED ICHTHYOSES AND ERYTHROKERATODERMAS | |||||||
|---|---|---|---|---|---|---|---|
| Diagnosis | Gene | Mode of inheritance | Incidence | Onset | Primary cutaneous features | Associated features | Diagnosis (in addition to molecular testing * ) |
| Common ichthyoses | |||||||
| Ichthyosis vulgaris | FLG | Autosomal semi-dominant | 1 : 100–1 : 250 | Infancy/childhood | Fine, adherent scales on extremities and trunk with sparing of flexures; larger scales on lower legs; hyperlinear palms/soles, furrowed heels | Keratosis pilaris; atopic dermatitis | Clinical; diminished/absent stratum granulosum; absent/reduced filaggrin immunostaining |
| Steroid sulfatase deficiency (X-linked recessive ichthyosis) | STS (deleted in ~90% of patients) | X-linked recessive | 1 : 2000–1 : 9500 boys/men | Infancy | Fine to large, dark, adherent scales on extremities, trunk, neck, and lateral face; sparing of skin folds | Corneal; opacities; cryptorchidism Female carriers : corneal opacities; prolonged labor with affected child | FISH or targeted/whole genome microarray to identify the gene deletion; increased plasma (hydroxy)cholesterol sulfate; lipoprotein electrophoresis (increased mobility of β-fraction); decreased steroid sulfatase activity in leukocytes |
| Nonsyndromic autosomal recessive congenital ichthyoses | |||||||
| Lamellar ichthyosis (LI), congenital ichthyosiform erythroderma (CIE) | LI > intermediate/CIE: TGM1 ABCA12 CYP4F22 SDR9C7 SULT2B1 ** CIE > intermediate/LI: ALOX12B ALOXE3 NIPAL4 / ICHTHYIN PNPLA1 CERS3 Childhood-onset intermediate: LIPN | Autosomal recessive † | LI: 1 : 200 000–1 : 300 000 CIE: 1 : 100 000–1 : 200 000 | Birth | Frequently collodion membrane at birth, then a generalized distribution; variable palm/sole involvement LI: large, thick, plate-like brown scales; absent or mild erythroderma CIE: fine, white scales; erythroderma | Heat intolerance; frequent (LI) or variable (CIE) scarring alopecia and ectropion > eclabium | Clinical; transglutaminase-1 immunostaining or in situ assay in skin biopsy specimen |
| Harlequin ichthyosis | ABCA12 (KDSR) | Autosomal recessive | Rare | Birth | Very thick, yellow–brown plates of scale that tightly encase the neonate; large, deep, bright red fissures; survivors develop severe CIE-like phenotype | Premature delivery; often neonatal death due to sepsis or respiratory insufficiency; extreme ectropion, eclabium, and ear deformities | Clinical |
| Keratinopathic ichthyoses (also includes ichthyosis with confetti – see text) | |||||||
| Epidermolytic ichthyosis | KRT1 KRT10 | Autosomal dominant (very rarely autosomal recessive) | 1 : 200 000–1 : 350 000 | Birth | At birth: erythroderma, blistering, erosions Later: hyperkeratosis with cobblestone pattern (most prominent over joints), ridging of the flexures; generalized or localized; variable degree of palmoplantar involvement and blistering | Frequent skin infections; malodor; gait and posture abnormalities | Clinical; histopathology |
| Superficial epidermolytic ichthyosis | KRT2 | Autosomal dominant | Rare | Birth | Erythroderma and blistering at birth; later, hyperkeratosis with flexural accentuation, “molting” of the skin; palms and soles spared | Clinical; histopathology | |
| Ichthyosis hystrix Curth–Macklin | KRT1 | Autosomal dominant | Rare | Birth | Mild to severe, mutilating palmoplantar keratoderma; hyperkeratosis with verrucous, cobblestone, or hystrix-like pattern on extremities and trunk | Pseudoainhum; digital contractures | Clinical; electron microscopy |
| Syndromic ichthyoses | |||||||
| Netherton syndrome | SPINK5 | Autosomal recessive | 1 : 50 000 | Birth/infancy | Congenital erythroderma and peeling; two principal phenotypes: ichthyosis linearis circumflexa and CIE-like; pruritus and eczematous plaques | Trichorrhexis invaginata and other hair shaft abnormalities; highly elevated serum IgE; neonatal temperature instability, electrolyte imbalance, failure to thrive; recurrent infections; food and other allergies; nonspecific aminoaciduria | Clinical; psoriasiform histopathology; trichoscopy, light microscopic hair shaft analysis; serum IgE measurement |
| Sjögren–Larsson syndrome | ALDH3A2 | Autosomal recessive | <1 : 100 000 | Birth | Fine to plate-like scaling or non-scaling hyperkeratosis; favors abdomen, neck, flexures; lichenification; pruritus | Spastic di- and tetraplegia; perifoveal glistening white dots; intellectual disability; white matter disease of the brain | Fatty aldehyde hydrogenase activity assay in cultured fibroblasts (limited availability) |
| Neutral lipid storage disease with ichthyosis | ABHD5 (CGI-58) | Autosomal recessive | Rare | Birth | Generalized fine, white scales with variable erythema | Developmental delay; hepatomegaly; myopathy; hearing impairment; cataracts; lipid-containing vacuoles in leukocytes | Peripheral blood smear to detect leukocyte lipid vacuoles; oil stains of frozen skin biopsy specimens |
| Trichothiodystrophy with ichthyosis | ERCC2 ERCC3 GTF2H5 Often not associated with ichthyosis or photosensitivity: MPLKIP/C7ORF11/TTDN1 RNF113A | Autosomal recessive X-linked | Rare | Birth | Generalized scaling with absent or minimal erythroderma, except during infancy; collodion membrane in ~30% of patients | Brittle hair and nails; photosensitivity in ~50% of patients; intellectual disability; gonadal abnormalities; short stature | Clinical; tiger tail pattern by microscopic hair shaft examination under polarizing light; analysis of hair sulfur/cysteine content |
| Erythrokeratodermas | |||||||
| Erythrokeratodermia variabilis | GJB3 GJB4 GJA1 (less common) | Autosomal dominant (very rarely autosomal recessive) | Rare | Birth/infancy | Transient, variable erythematous patches; more stable, geographic, hyperkeratotic plaques over knees, elbows, Achilles tendons, extremities, buttocks, lateral trunk; face and scalp often spared; less common generalized hyperkeratosis; palmoplantar keratoderma in ~50% of patients | Burning or stinging sensation preceding or accompanying erythema | Clinical, particularly history or presence of transient erythema |
| Progressive symmetric erythrokeratoderma | No consistently implicated genes ( LOR ) ( GJB4 ) ( KRT83 ) ( KDSR – see Table 57.5 ) | Autosomal dominant Autosomal recessive | Rare | Infancy/childhood | Fixed, slowly progressive, erythematous, hyperkeratotic plaques with sharp, figurate borders; on cheeks, over knees, elbows, extremities, rarely trunk; palmoplantar keratoderma common | Clinical | |
| Keratitis–ichthyosis–deafness syndrome | GJB2 (GJB6 – single reported patient) | Autosomal dominant | Rare | Birth/infancy | Transient neonatal erythroderma; erythematous, hyperkeratotic plaques with well-demarcated, borders on face and extremities; follicular keratoses; thickening of the skin with an appearance of “coarse-grained leather”; stippled palmoplantar keratoderma | Congenital sensorineural hearing impairment; progressive keratitis with corneal neovascularization, conjunctivitis; recurrent mucocutaneous infections, especially with Candida albicans ; increased susceptibility to oral and cutaneous squamous cell carcinoma; nail, hair and dental anomalies; very rare fatal form with respiratory problems | Clinical |
| Syndromic X-linked dominant ichthyosiform conditions | |||||||
| CHILD syndrome | NSDHL | X-linked dominant | Rare | Birth | At birth, unilateral erythema and waxy, yellowish adherent scale; later, verrucous; hyperkeratosis of variable extent, with affinity for skin folds | Ipsilateral skeletal hemidysplasia, organ hypoplasia | Clinical |
| Conradi–Hünermann–Happle syndrome | EBP | X-linked dominant | Rare | Birth | At birth, ichthyosiform erythroderma with feathery, adherent scale along lines of Blaschko; later, follicular atrophoderma along lines of Blaschko | Unilateral cataracts; chondrodysplasia punctata (during infancy); asymmetric skeletal abnormalities; patchy scarring alopecia | Clinical; epiphyseal stippling on X-rays during infancy; accumulation of plasma 8(9)cholesterol |
* Genetic testing is available for all types of ichthyosis with a known molecular basis through clinical or research laboratories; multigene panels are preferable over single gene testing if the diagnosis is not evident based on clinical and laboratory findings.
** Distribution similar to that of steroid sulfatase deficiency, with sparing of the central face and skin folds.
† There are also rare reports of autosomal dominant inheritance of LI or CIE phenotypes (occasionally with a collodion membrane at birth) with unknown genetic bases. Biallelic small deletions in the caspase 14 gene ( CASP14 ) were recently identified in patients from two Algerian families who presented with generalized fine whitish scales with no erythema and no collodion membrane.
Milestones in recognizing distinct entities include the description of harlequin ichthyosis in the nineteenth century, separation of the bullous and non-bullous types of ichthyosis by Brocq (who also coined the name “congenital ichthyosiform erythroderma”) in the early 20th century , and differentiation between autosomal dominant “epidermolytic hyperkeratosis” and autosomal recessive “lamellar ichthyosis” by Frost and Van Scott in 1966 . Alibert mentioned the autosomal dominant inheritance of ichthyosis vulgaris as early as 1806 . X-linked ichthyosis, despite several earlier reports, was only fully recognized in Wells and Kerr’s study in 1966 , followed by the identification of steroid sulfatase deficiency in the 1970s . Advances in the molecular etiology and biology of ichthyoses and erythrokeratodermas have provided tools to categorize these disorders of cornification, at least in part, based on their underlying genetic defects. In 2009, an international group of clinical and research experts developed a revised nomenclature and classification system for ichthyoses and other disorders of cornification, incorporating molecular causes as well as functional aspects of disease pathogenesis .
Establishing the correct diagnosis in a patient with ichthyosis is a prerequisite for making prognostic predictions, planning therapy, and offering genetic counseling. This may pose a considerable challenge, as these disorders are uncommon and have overlapping clinical spectra. However, a systematic approach utilizing clinical and laboratory-based clues can aid in making the diagnosis ( Fig. 57.1 ).
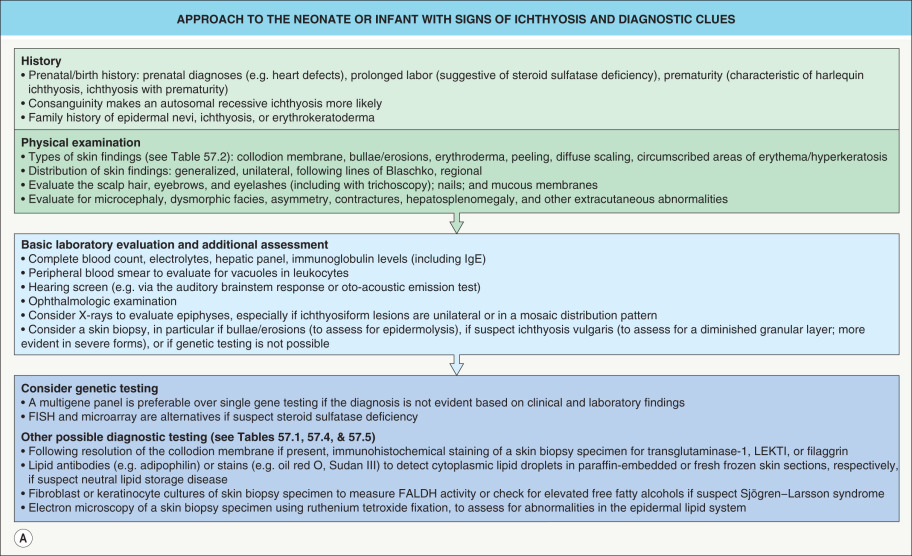
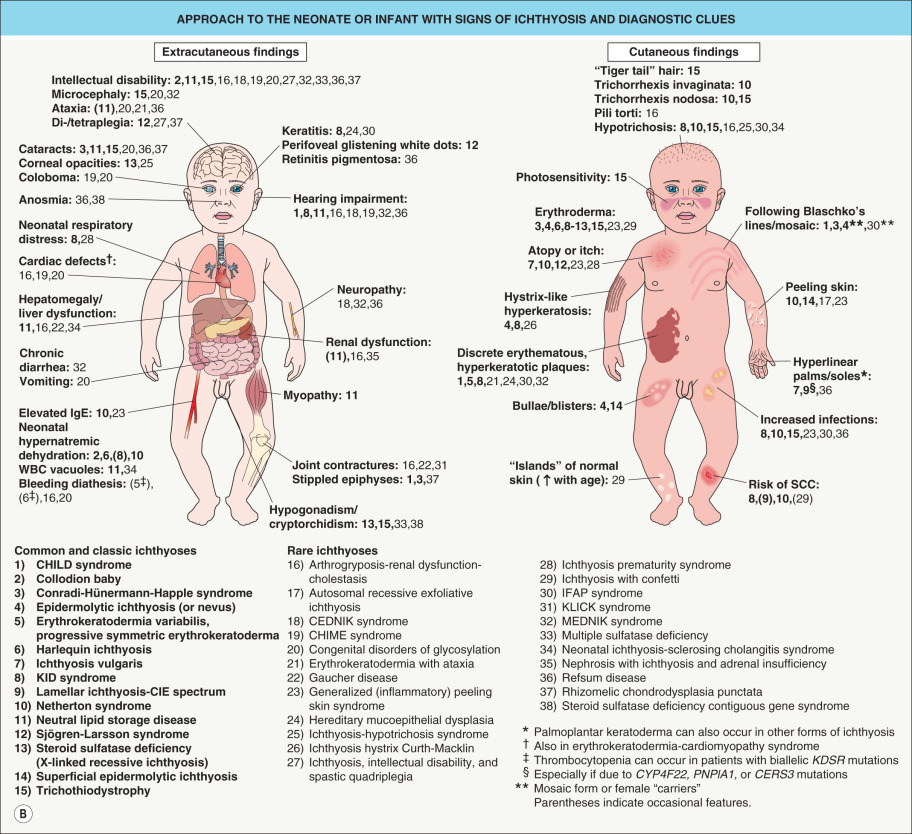
In general, it is helpful to determine whether an ichthyosis presented at birth (e.g. as a collodion baby) or later in life and whether manifestations are limited to the skin or are part of a multisystem disorder. The quality and distribution of the scale as well as the presence or absence of erythroderma, blistering, and abnormalities of cutaneous adnexa are other useful clinical features. A thorough family history is essential for recognizing the inheritance pattern, and examination of both parents (even in a seemingly sporadic case) may reveal valuable diagnostic hints such as an epidermal nevus representing a mosaic presentation of epidermolytic ichthyosis. Patients whose parents are clinically unaffected may have: (1) a recessive ichthyosis, especially in the setting of consanguinity and/or affected siblings; (2) a dominant ichthyosis due to a “new” mutation; or (3) for a male patient, an X-linked recessive ichthyosis where the mother may be an asymptomatic carrier but her male relatives could potentially be affected. A few disorders can be recognized based on characteristic histopathologic and ultrastructural features, such as epidermolytic ichthyosis and ichthyosis hystrix Curth–Macklin. Laboratory studies suggest the diagnosis in other conditions, including enzyme analysis in steroid sulfatase deficiency and observation of lipid vacuoles in circulating leukocytes in neutral lipid storage disease (see Fig. 57.1 ).
This strategy allows the clinician to identify many ichthyoses based on their key features. For most conditions, the molecular etiology is known and genetic testing can confirm the diagnosis, allow more precise genetic counseling, and provide the basis for prenatal testing. However, despite advances in our understanding of the pathomechanisms underlying ichthyoses, effective therapies are available for only a small subset of these conditions. Hopefully, ongoing research will lead to the development of targeted treatments that will be of greater benefit to ichthyosis patients. Patient organizations such as the Foundation for Ichthyosis and Related Skin Types (FIRST; www.firstskinfoundation.org ) can provide valuable support to affected individuals and their families.
Ichthyoses
Ichthyosis Vulgaris
▪ Ichthyosis simplex ▪ Autosomal dominant ichthyosis
History
In 1806, Alibert described “ichthyose nacrée”, which represented the first well-documented report of ichthyosis vulgaris. In 1966, Wells and Kerr separated autosomal dominant and X-linked recessive forms of ichthyosis and delineated their clinical and epidemiological features.
Epidemiology
Ichthyosis vulgaris is the most common disorder of cornification, with an estimated prevalence of ~1 in 100 to 1 in 250 individuals . It is inherited in an autosomal semi-dominant manner, with mild ichthyosis in individuals with heterozygous filaggrin mutations and more severe ichthyosis in those with complete filaggrin deficiency due to mutations on both alleles . The overall penetrance in heterozygotes is ~80–95% for ichthyosis vulgaris and/or associated findings (e.g. hyperlinear palms, keratosis pilaris, atopic dermatitis) , although there is variable phenotypic expression between and within families.
Pathogenesis
Ichthyosis vulgaris is caused by loss-of-function mutations in the filaggrin gene ( FLG ) (see Fig. 56.9 ) . Approximately 8% of European and 3% of East Asian individuals harbor at least one FLG mutation, compared to <1% among those of Mediterranean or African descent . R501X and c.2282del4 account for up to 80% of mutations in Northern Europeans, while different null mutations are common in other population groups.
Profilaggrin represents a major component of the keratohyalin granules located in the granular layer of the epidermis. During keratinocyte differentiation, profilaggrin is cleaved into filaggrin peptides, which aggregate keratin intermediate filaments. These complexes are cross-linked to the cornified cell envelope and are responsible for proper formation of compact squamous cells (see Fig 56.1 , Fig 56.2 ). Filaggrin is eventually degraded into water-retaining amino acids, in particular histidine, that serve as a natural moisturizer. Filaggrin deficiency results in impaired cornification, increased transepidermal water loss, enhanced penetration of allergens and irritants, and a propensity to develop inflammatory responses upon exposure to the latter. This explains the association of FLG mutations with atopic dermatitis (see Ch. 12 ) as well as hand eczema, irritant contact dermatitis, and nickel contact sensitization . In addition, an association between FLG mutations and elevated serum vitamin D levels may result from increased UVB-induced vitamin D production in the skin, perhaps related to a paucity of urocanic acid, a photoprotective histidine metabolite. The flaky-tail mouse with biallelic loss-of-function mutations in Flg represents an animal model that serves as a tool for the study of ichthyosis vulgaris and other filaggrin-deficient conditions.
Clinical Features
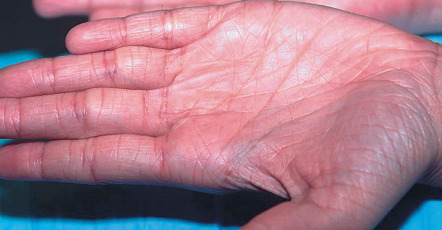
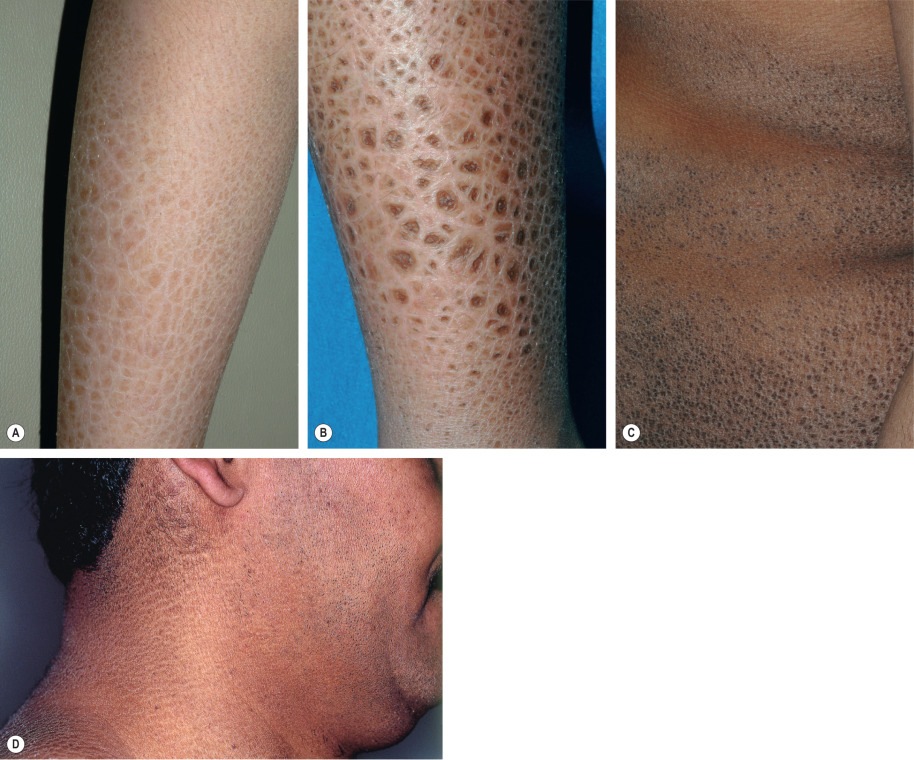
Ichthyosis vulgaris in FLG heterozygotes is usually not evident at birth. Dry skin and mild to moderate scaling appear during infancy or early childhood. Fine, white, flaky scales typically develop on the extremities, especially the extensor surfaces ( Fig. 57.2 ). The groin and flexural areas are spared because of increased humidity in those regions. On the lower legs, the scales are often larger with an adherent center and detached, outward-turning edges. Mild hyperkeratosis of the palms and soles leading to accentuated skin markings (hyperlinearity) is common ( Fig. 57.3 ).
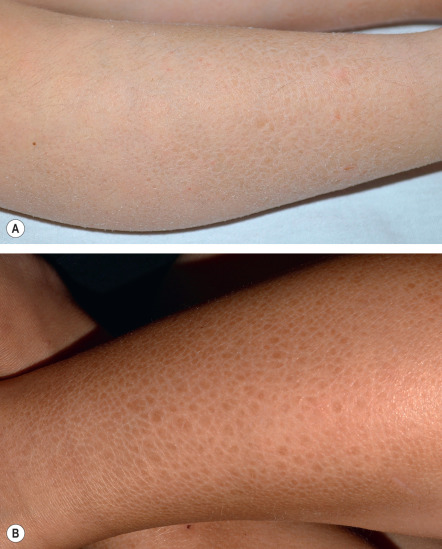
In more severe disease due to a complete lack of filaggrin, ichthyosis vulgaris may present at birth with mild erythema and generalized scaling or peeling. Later, large scales reminiscent of lamellar ichthyosis may develop and the trunk, scalp, forehead, and cheeks can be affected in addition to the extremities. Palmoplantar involvement is also more pronounced and often results in furrows or painful fissures of the heels.
Severity and symptoms such as pruritus depend on the season and climate, improving during the summer and with higher humidity, and worsening in a dry, cold environment. Although the ichthyosis may be progressive during childhood, it usually improves with age. Ichthyosis vulgaris is frequently associated with keratosis pilaris and the atopic triad of asthma, hay fever, and atopic dermatitis. The latter is encountered in at least 25–50% of patients with ichthyosis vulgaris and can obscure the characteristic sparing of the flexures. Conversely, 10–15% of all individuals with atopic dermatitis and up to 50% of those of Northern European descent with atopic dermatitis also have moderate to severe ichthyosis vulgaris .
Pathology
Mild orthokeratotic hyperkeratosis is seen and a diminished granular layer is often but not always observed ( Fig. 57.4A ). Approximately 30–50% of affected individuals have no detectable granular layer and no keratohyalin granules by electron microscopy , while others have structural abnormalities in keratohyalin granules. Immunohistochemistry demonstrates diminished or absent filaggrin staining in most patients ( Fig. 57.4B & C ).
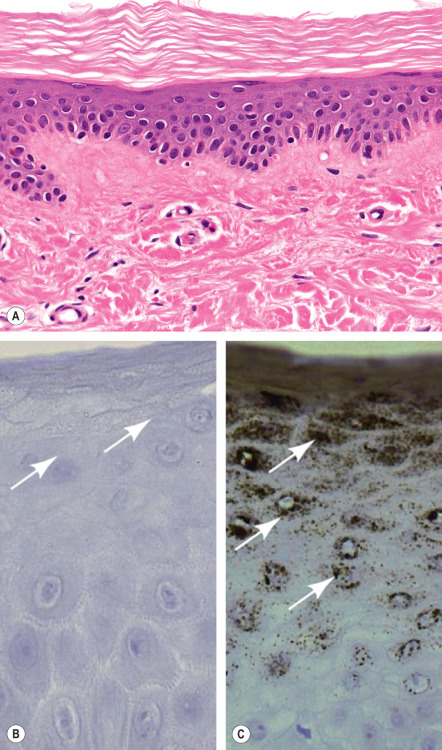
Differential Diagnosis
The border between dry skin (xerosis) and mild ichthyosis vulgaris is not clear-cut. In male patients, X-linked ichthyosis may be differentiated by larger, darker scales and involvement of the neck and other flexures; cryptorchidism, a maternal history of delayed or prolonged labor, and the inheritance pattern can represent additional clues. Chromosomal microarray or fluorescence in situ hybridization (FISH) testing can exclude steroid sulfatase deficiency. Acquired ichthyosis is distinguished by its development later in life and association with conditions such as malnutrition, infections (e.g. leprosy), neoplasms (e.g. lymphoma), and inflammatory disorders (e.g. sarcoidosis). Some individuals with other heritable forms of ichthyosis also have a partial or complete filaggrin deficiency, which may result in a more severe phenotype than is usually observed for those ichthyoses.
Treatment
The mainstay of treatment is application of emollients, humectants, and keratolytic agents to reduce scaling. Preparations that contain urea and/or glycolic, lactic, or salicylic acid are usually beneficial; with the latter, care must be taken to prevent salicylate toxicity (see Ch. 129 ). Skin irritation from topical retinoids limits their use for ichthyosis vulgaris, and vitamin D analogues are typically ineffective. Systemic treatment with acitretin or isotretinoin is rarely indicated. The use of moisturizing cleansers and humidifiers may also be helpful. Research is underway to identify compounds that could upregulate filaggrin expression and, for those patients with nonsense mutations, modify protein translation .
Steroid Sulfatase Deficiency
▪ X-linked recessive ichthyosis
History
A type of ichthyosis occurring only in boys and men was first reported in the early nineteenth century, and its X-linked recessive inheritance was clearly recognized in the 1920s. In 1966, the clinical features of this entity were distinguished from those of ichthyosis vulgaris . The underlying steroid sulfatase deficiency was identified in the late 1970s.
Epidemiology
The worldwide incidence is between 1 in 2000 and 1 in 9500 male births. It is an X-linked recessive disorder that is transmitted by asymptomatic female carriers and almost exclusively affects boys and men.
Pathogenesis
Diminution or complete absence of steroid sulfatase activity is caused by a deletion of the entire STS gene on chromosome Xp22.31 in ~90% of patients and inactivating mutations in others . Steroid sulfatase deficiency results in impaired hydrolysis of cholesterol sulfate and dehydroepiandrosterone sulfate (DHEAS), with subsequent accumulation of cholesterol 3-sulfate in the epidermis. High levels of this metabolite can inhibit transglutaminase-1, explaining the partial overlap with lamellar ichthyosis.
In women pregnant with an affected fetus, steroid sulfatase deficiency in the fetal placenta causes low or absent levels of estrogen in the urine and amniotic fluid because of inadequate deconjugation of DHEAS, which is necessary for estrogen synthesis. As a result, labor often fails to initiate spontaneously or progress, due to insufficient dilation of the cervix. This can only be partially overcome by oxytocin administration, often necessitating cesarean section.
Clinical Features
In ~90% of affected boys, steroid sulfatase deficiency presents during the neonatal period with mild erythroderma and generalized peeling, often with exfoliation of large, translucent scales. The typical polygonal, dark-brown, adherent scales develop later during infancy or childhood and are distributed symmetrically on the extremities, trunk, and neck ( Fig. 57.5 ). The scales are occasionally grayish or white, and they tend to be larger on the lower extremities. Preauricular involvement is highly characteristic and the neck is almost invariably affected, which has resulted in the descriptive term “dirty neck disease” (see Fig. 57.5D ); other flexural areas may or may not be involved. Fine scaling of the scalp is frequently seen during early childhood but diminishes over time. The palms, soles, and remainder of the face are characteristically spared. The ichthyosis tends to improve in the summer; however, in contrast to ichthyosis vulgaris, it does not significantly subside with age.
Asymptomatic corneal opacities occur in 10–50% of male patients and in some female carriers, but other ocular abnormalities such as deuteranopia (green color-blindness) are rare. Male patients have a 20-fold increased incidence of cryptorchidism and, independent of testicular maldescent, may have a slightly increased risk for developing testicular cancer and hypogonadism . A higher prevalence of attention deficit hyperactivity disorder has also been reported . In addition, ~5% of patients with steroid sulfatase deficiency have a larger contiguous gene deletion, which may include the genes that underlie Kallmann syndrome (hypogonadotropic hypogonadism with anosmia), X-linked chondrodysplasia punctata, ocular albinism, intellectual disability/autism, and short stature.
Steroid sulfatase activity is measurably reduced in 85% of female carriers, but the remaining activity is sufficient to prevent skin manifestations. Because the STS gene at least partially escapes X-chromosome inactivation, mosaic ichthyotic changes are not seen.
Pathology
Histopathologic findings include hyperkeratosis or parakeratosis overlying a normal or slightly thickened granular layer. Follicular hyperkeratosis may be present. Electron microscopy shows an increased size and number of keratohyalin granules. In the stratum corneum, desmosomes are retained and cells contain a large number of melanosomes. Biochemical and cell kinetic studies show normal epidermal cell turnover and water homeostasis.
Other diagnostic tests
Molecular analysis via targeted or whole genome microarrays, FISH, and PCR assays is available through diagnostic laboratories and is useful for detecting the underlying genetic defect, which, when known in a female carrier, may also be utilized for prenatal diagnosis via chorionic villi or amniotic fluid samples. However, non-invasive prenatal diagnosis through decreased maternal serum estriol levels and the presence of non-hydrolyzed sulfated steroids in maternal urine is also possible. Accumulation of (hydroxy)cholesterol sulfate can be detected indirectly by increased migration of the β-fraction in serum lipoprotein electrophoresis, or measured directly by liquid chromatography-mass spectrometry (LC-MS) of serum, epidermal scale, placental tissue, or amniotic fluid. In addition, a biochemical assay can measure steroid sulfatase enzymatic activity in leukocytes, fibroblasts, keratinocytes, or placental tissue.
Differential Diagnosis
Clinically, ichthyosis vulgaris is distinguished by its sparing of the neck and other flexural areas as well as the presence of hyperlinear palms and keratosis pilaris. As discussed above, an identical ichthyosis phenotype can be associated with extracutaneous findings in patients with larger deletions encompassing contiguous genes. Refined karyotyping, FISH, or microarray analysis can be utilized to detect Xp deletions and X;Y translocations in patients who present with these additional findings. Autosomal recessive congenital ichthyosis due to defective cholesterol sulfotransferase ( SULT2B1 ) has a phenotype similar to that of steroid sulfatase deficiency, with lamellar scaling that spares the central face and skin folds .
Treatment
Topical humectants (especially propylene glycol), keratolytics, and retinoids are helpful alone or in combination. In contrast, vitamin D analogues are less effective and cause significant irritation. Systemic retinoids are rarely necessary.
Epidermolytic Ichthyosis
▪ Bullous congenital ichthyosiform erythroderma (Brocq) ▪ Epidermolytic hyperkeratosis ▪ Bullous ichthyosis
History
Nikolsky first recognized the characteristic histopathology of the bullous form of congenital ichthyosis in 1897, and Brocq differentiated between dry (non-bullous) and wet (bullous) forms of congenital ichthyosiform erythroderma in 1902 . Based on the distinctive histopathologic features of the epidermis, Frost and Van Scott introduced the name “epidermolytic hyperkeratosis” for this autosomal dominant blistering form of congenital ichthyosis in 1966.
Epidemiology
Epidermolytic ichthyosis (EI) has an estimated worldwide prevalence of 1 in 200 000 to 1 in 350 000. It is generally an autosomal dominant disorder with complete penetrance, with rare reports of autosomal recessive inheritance. Both genders are affected equally. At least 50% of all cases occur sporadically due to new mutations.
Pathogenesis
EI is caused by heterozygous mutations in the genes encoding keratin 1 (KRT1) and keratin 10 (KRT10) . Very rarely, in consanguineous families, a severe autosomal recessive form of EI has been described that is due to loss-of-function mutations . Keratins 1 and 10 are coexpressed in the differentiated spinous and granular layers of the epidermis, which are the sites of disease pathology in this disorder. KRT1 mutations are usually associated with severe palmoplantar keratoderma, whereas most KRT10 mutations result in sparing of the palms and soles because there is less expression of KRT10 in these locations . Pathogenic mutations alter highly conserved amino acid residues located at the boundaries of the α-helical rod region; these sites represent mutational “hot spots” that impact the helix initiation and termination motifs (see Fig. 56.5 ) . Mutations in other sites are uncommon and generally associated with a milder or unusual phenotype .
Mutations perturb keratin alignment, oligomerization, and filament assembly; this weakens the cytoskeleton, compromises the mechanical strength and cellular integrity of the epidermis, and leads to cytolysis and blistering. Epidermal acanthosis and hyperkeratosis result from hyperproliferation, decreased desquamation, and other factors. The barrier function of the skin is markedly disturbed, leading to increased transepidermal water loss and bacterial colonization of the stratum corneum.
Clinical Features
EI presents at birth with erythroderma ( Fig. 57.6A ), peeling, erosions, and often widespread areas of denuded skin (see Fig. 34.16 ). Hyperkeratosis may be present, but it often develops later during infancy. Skin fragility, blistering, and erythema decrease over time, and severe hyperkeratosis prevails ( Fig. 57.6B–E ). However, patients may still periodically shed large plates of superficial epidermis, revealing a tender, erythematous base. Ridges along skin lines are common in flexures (see Fig. 57.6C, D ), while hyperkeratosis over the extensor surfaces of the joints often forms a cobblestone pattern (see Fig. 57.6E ). When present, palmoplantar keratoderma may be severe and associated with digital contractures ( Fig. 57.6F ).
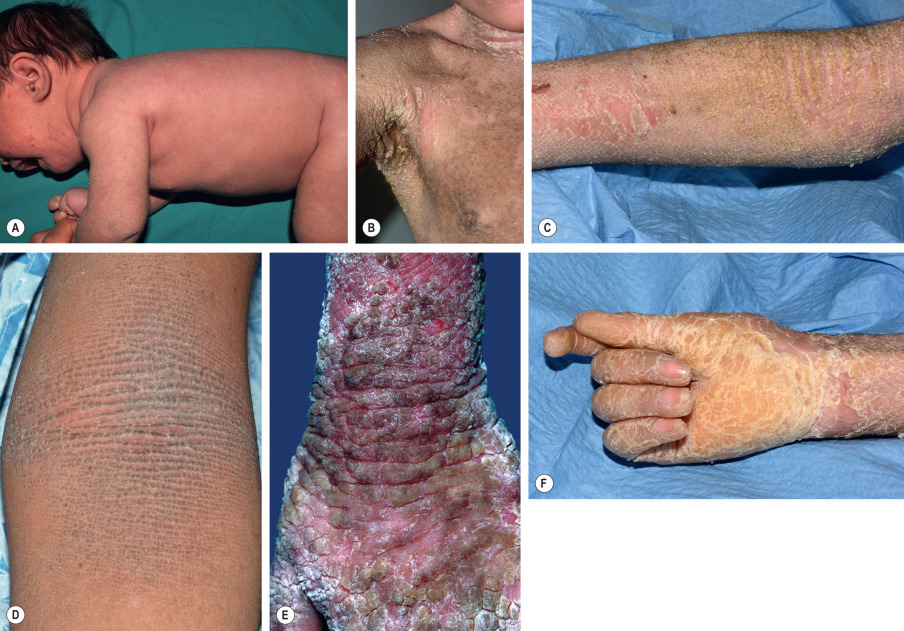
The clinical presentation varies considerably among patients and families, and six characteristic clinical patterns with or without palmoplantar involvement have been described . Unusual clinical variants of EI include “cyclic ichthyosis with epidermolytic hyperkeratosis” and “annular epidermolytic ichthyosis”, which are caused by distinct mutations outside the helix boundaries of KRT1 and KRT10 , respectively .
EI is disfiguring and has tremendous impact on patients’ quality of life and social interactions. Sepsis as well as fluid and electrolyte imbalances can be life-threatening in the neonatal period. Episodes of blistering and secondary skin infections are also problematic later in life. The disorder is accompanied by a pungent body odor, and it is occasionally associated with posture and gait abnormalities. Angular cheilitis and severe scalp involvement leading to encasement of hair shafts and hair loss are additional associated findings.
Epidermolytic epidermal nevi (mosaic epidermolytic ichthyosis)
The mosaic form of EI is characterized by unilateral or bilateral streaks of hyperkeratosis that follow the lines of Blaschko . Extensive involvement with marked hyperkeratosis leading to protruding, porcupine-like spines has been referred to as “ichthyosis hystrix”, but this is a descriptive term rather than the name of a distinct disorder. Epidermolytic epidermal nevi are caused by a postzygotic mutation in KRT1 or KRT10 that arises during embryogenesis. If this mutation involves gonadal cells, it can potentially be transmitted to the patient’s offspring, resulting in full-blown, generalized disease . Examination of both parents of a child with sporadic EI is therefore recommended.
Pathology
Highly characteristic structural and ultrastructural abnormalities described as “epidermolytic hyperkeratosis” allow EI to be distinguished from other congenital ichthyoses. Key features on routine histologic examination are dense orthokeratotic hyperkeratosis, prominent acanthosis, hypergranulosis, and cytolysis of the suprabasal and granular layers leading to small intraepidermal blisters ( Fig. 57.7 ). Keratinocytes exhibit marked intracellular vacuolization and dense clumps of keratin intermediate filaments (KIFs). A mild perivascular lymphohistiocytic infiltrate is usually present in the upper dermis. Ultrastructural analysis reveals fragmented, clumped KIFs in the lower epidermis and perinuclear KIF shells in the upper epidermis.
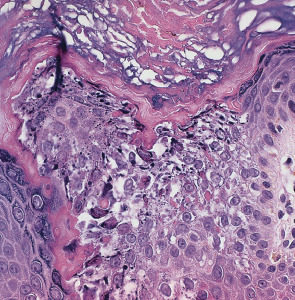
Other diagnostic tests
Mutation screening and complete sequencing of KRT1 and KRT10 is commercially available. Prenatal diagnosis can be performed when the underlying mutation has been identified in affected family members.
Differential Diagnosis
In the neonatal period, the presence of blisters and erosions differentiates EI from non-bullous congenital ichthyoses ( Table 57.2 ). Clinical distinction from the various forms of epidermolysis bullosa, staphylococcal scalded skin syndrome, and other vesiculobullous and erosive disorders that can present in neonates may require skin biopsy specimens and cultures (see Chs 32 & 34).
| NEONATAL PRESENTATION OF SELECTED ICHTHYOSES AND RELATED DISORDERS | |||||
|---|---|---|---|---|---|
| Disorder | Collodion membrane | Bullae/blisters/erosions | Erythroderma | Generalized ichthyosis | Peeling/exfoliation |
| Epidermolytic ichthyosis | + | + | (+) | (+) | |
| Superficial epidermolytic ichthyosis | (+) | (+) | (+) | + | |
| Lamellar ichthyosis | + | (+) | + | ||
| Congenital ichthyosiform erythroderma | + | + | + | ||
| Self-improving collodion ichthyosis | + | ||||
| Sjögren–Larsson syndrome | (+) | + | + | ||
| Neutral lipid storage disease | (+) | + | + | ||
| Trichothiodystrophy | (+) | + | + | ||
| Netherton syndrome | + | + | + | ||
| Steroid sulfatase deficiency | + | (+) | + | ||
| Peeling skin syndromes (see Table 57.5 ) | (+) | (+) | + | ||
| Conradi–Hünermann–Happle syndrome | (+) | + (lines of Blaschko) | + (lines of Blaschko) | ||
| CHILD syndrome | Unilateral | Unilateral * | |||
| KID syndrome | + | (+) | |||
* Some attenuated or band-like involvement may occasionally be appreciated on the contralateral side.
Later in life, EI can be distinguished from other congenital ichthyoses by the history of blistering at birth with focal recurrences, frequent superinfections, characteristic clinical appearance, and distinctive histologic features. The latter may also be seen in epidermolytic palmoplantar keratoderma (see Ch. 58 ), which is limited to the palms and soles. Superficial EI (ichthyosis bullosa of Siemens) lacks erythroderma and shows “molting” of the superficial epidermis due to epidermolysis within the granular layer. Ichthyosis hystrix Curth–Macklin sometimes resembles EI, with ridged, verrucous hyperkeratosis over joints and in flexures (see below). In contrast to EI, there is no clinical or histologic evidence of either blister formation or epidermolysis, and specific ultrastructural abnormalities of the KIFs are present.
Treatment
Treatment is symptomatic and depends on the age of the patient and clinical issues. In the neonatal period, infants require management in an intensive care nursery to provide protective isolation and prevent or treat dehydration, electrolyte imbalance, and cutaneous superinfection. Sepsis should be treated with broad-spectrum antibiotics. When the neonate is handled carefully and protective padding and lubricants are used, erosions and denuded skin usually heal rapidly.
In children and adults, therapy is aimed at reducing hyperkeratosis, removing scale, and softening the skin. Keratolytic creams and lotions containing urea, salicylic acid, and α-hydroxy acids are effective but often not well tolerated, especially in children, because of burning and stinging. Widespread topical application of higher-concentration salicylic acid preparations should be avoided because of the risk of systemic salicylism. Topical tretinoin and vitamin D preparations may be beneficial but often cause skin irritation. Frequent use of emollients and humectants can be combined with hydration (e.g. soaking of the skin during bathing) and mechanical abrasion of keratotic skin via gentle scrubbing with a soft brush or sponge.
Bacterial skin infections are common and often trigger blistering; they require topical or systemic antibiotic treatment. Use of antiseptics such as antibacterial soaps, chlorhexidine, or dilute sodium hypochlorite baths may help to control bacterial colonization. Continuous use of oral or topical antibiotics should be avoided due to the risk of developing antibiotic resistance. Because of the increased skin fragility, it is also important to prevent mechanical trauma, e.g. by wearing comfortable clothing and shoes. Oral retinoids may drastically reduce hyperkeratosis and the frequency of infections in patients with generalized EI, but they also increase epidermal fragility and blistering. Low initial doses with gradual increase and careful monitoring are advisable, with use of the lowest effective maintenance dose . The disfigurement and malodor often affect psychosocial functioning.
Superficial Epidermolytic Ichthyosis
▪ Ichthyosis bullosa of Siemens ▪ Ichthyosis exfoliativa
History
In 1937, Siemens first recognized this congenital ichthyosis as a distinct entity and differentiated it from classic EI.
Pathogenesis
Superficial EI is a rare autosomal dominant ichthyosis caused by heterozygous mutations in the keratin 2 gene ( KRT2 ), which is expressed only in the uppermost spinous and granular cell layers of the epidermis. Mutations cluster at the boundaries of the rod domain of KRT2; Glu487Lys within the helix termination motif is most frequently identified .
Clinical Features
The phenotype is milder than in classic EI. At birth, the skin may appear normal or show mild blistering. Trauma-induced small blisters on the extremities occur during infancy but usually subside by early childhood, while hyperkeratosis develops. Predilection sites are the skin overlying the joints, the flexures, and the dorsa of the hands and feet ( Fig. 57.8 ), always sparing the palms and soles. The skin may appear ridged, shiny, or lichenified. A characteristic feature is superficially denuded areas with collarette-like borders, described as “molting” or “Mauserung”, which develop due to superficial blistering and shedding of the stratum corneum.
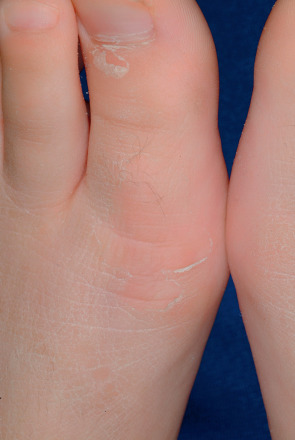
Pathology
Histopathologic abnormalities include orthokeratotic hyperkeratosis, acanthosis, and vacuolization of the granular cell layer that occasionally leads to superficial intraepidermal separation. By electron microscopy, clumping of tonofilaments is present but limited to the upper spinous and granular cell layers.
Differential Diagnosis
Infants usually do not present with the marked erythroderma and widespread blistering seen in classic EI, and later in life there is a milder course with more superficial skin shedding and a lack of erythema. Peeling skin syndromes can have a similar clinical appearance but do not feature vacuolization of the granular layer. Recurrent blistering is also a feature of epidermolysis bullosa simplex, but this disorder does not have associated hyperkeratosis other than an occasional palmoplantar keratoderma and can be differentiated based on deeper blisters due to basal epidermolysis.
Treatment
The management is similar to that of EI.
Ichthyosis With Confetti
▪ Ichthyosis en confetti ▪ Confetti ichthyosis ▪ Congenital reticular ichthyosiform erythroderma ▪ Ichthyosis variegata
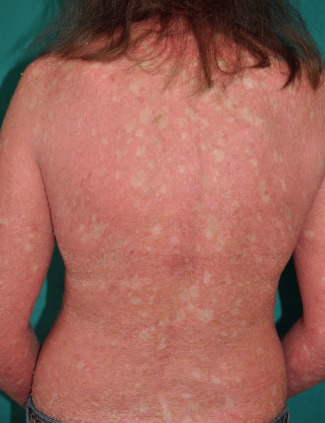
This ichthyosis is caused by heterozygous frameshift mutations in KRT10 or less often KRT1 . The resulting mutant keratin 10 protein has an arginine-rich carboxy terminus, which redirects the protein from its normal cytoplasmic location to the nucleolus. Each island of normal skin represents a revertant clone that arises via mitotic recombination, i.e. a “crossing-over” process that leads to homozygous mutant and homozygous normal daughter cells. The exact role of the frameshift mutations in the markedly elevated rate of mitotic recombination remains to be determined.
Ichthyosis Hystrix Curth–Macklin
History
In 1954, Ollendorff-Curth and Macklin described a family with hystrix-like hyperkeratosis and palmoplantar keratoderma. Electron microscopic examinations by Anton-Lamprecht et al. revealed peculiar ultrastructural abnormalities of the keratinocyte cytoskeleton. Since the initial description, a few large families with autosomal dominant inheritance and sporadic cases have been reported.
Pathogenesis
In a few unrelated patients with ichthyosis hystrix Curth–Macklin, heterozygous mutations in the keratin 1 gene ( KRT1 ) have been demonstrated. In contrast to EI, the mutations were small nucleotide deletions/insertions that led to a frameshift and produced a keratin 1 protein with an aberrant tail. Because the latter lacks the usual glycine loop motifs, protein–protein interactions are altered .
Clinical Features
The hyperkeratotic areas in ichthyosis hystrix Curth–Macklin can clinically mimic EI, but there is no skin fragility. Clinical expression varies even within families, ranging from isolated palmoplantar keratoderma (which may be severe and mutilating) to localized hyperkeratotic plaques to generalized hystrix-like hyperkeratosis with horny, rigid spikes. Hyperkeratosis is present at birth or develops during early childhood. It has a cobblestone-like or ridged surface over large joints and in skin folds. Circular constriction bands (pseudoainhum), starfish-like keratoses, knuckle pads, digital flexion contractures, and secondary bacterial infections have been described.
Pathology
Most histopathologic features, including orthokeratotic hyperkeratosis, hypergranulosis, acanthosis, and church spire-like papillomatosis, are nonspecific. Cells in the differentiated cell layers may be vacuolated or binucleated. Ultrastructural abnormalities in the upper epidermis are diagnostic, with a shell-like, interspersed network of tangled KIFs, often associated with perinuclear vacuolization and formation of binucleated cells. In contrast to EI, there is no epidermolysis or keratin clumping.
Differential Diagnosis
Ichthyosis hystrix Curth–Macklin can be differentiated from EI and epidermolytic palmoplantar keratoderma by the absence of blistering and skin fragility as well as by electron microscopic findings.
Treatment
The most effective treatments are systemic retinoids and topical keratolytic agents (see EI above).
Ichthyosis Hystrix
▪ “Porcupine man” ▪ Ichthyosis hystrix gravior type Lambert ▪ Ichthyosis hystrix gravior type Rheydt (the initial term for hystrix-like ichthyosis with deafness [HID] syndrome) ▪ Systematized verrucous nevus
Collodion Baby
▪ Collodion fetus ▪ Ichthyosis congenita ▪ Self-improving collodion ichthyosis – self-healing collodion baby, lamellar exfoliation of the newborn, desquamation of the newborn, self-improving collodion baby
Epidemiology
The collodion baby is a shared presentation of several congenital ichthyoses, most with autosomal recessive inheritance. It is not a distinct disorder.
Pathogenesis
The pathomechanisms leading to formation of a collodion membrane are not completely understood, but this presentation points to disturbed adaptation of the fetal epidermis to postnatal life. Collodion babies who eventually develop lamellar ichthyosis have been shown to harbor mutations in the transglutaminase-1 gene, leading to deficiency of this crucial cross-linking enzyme of the epidermis . Transglutaminase-1 deficiency perturbs formation of the cornified cell envelope, resulting in hyperkeratosis, profoundly impaired barrier function, and transepidermal water loss. Defects in multiple lipid-processing enzymes or lipid transporters have been demonstrated in other congenital ichthyoses with a collodion membrane presentation, such as lipoxygenase deficiency in congenital ichthyosiform erythroderma, ABCA12 deficiency in lamellar ichthyosis, fatty aldehyde dehydrogenase deficiency in Sjögren–Larsson syndrome, deficiency of an activator of the adipose triglyceride lipase in neutral lipid storage disease, and β-glucocerebrosidase deficiency in Gaucher disease (see Fig. 56.2 ). These findings underscore the importance of lipids to epidermal barrier function.
In ~10% of collodion babies , a milder “self-healing” phenotype known as self-improving collodion ichthyosis occurs. This has been reported in association with TGM1, ALOX12B, ALOXE3 , NIPAL4 (ICHTHYIN) , or CYP4F22 mutations that fully inactivate the encoded protein in utero but not after birth . It has been postulated that this “dynamic” phenotype is dependent on environmental conditions which influence the stability of the mutant protein. A form of self-improving collodion ichthyosis in which membranes are localized to acral areas due to compound heterozygous TGM1 mutations has also been described .
Clinical Features
Collodion babies are often born prematurely and have increased perinatal morbidity and mortality. At birth, the neonate is covered with a taut, shiny, transparent membrane formed by the thickened stratum corneum, which resembles a plastic wrap ( Fig. 57.10 ). Its tautness often leads to ectropion, eclabium, and hypoplasia of nasal and auricular cartilage. Sucking and pulmonary ventilation may also be impaired, resulting in dehydration, malnutrition, hypoxia, and pneumonia. After birth, the parchment-like membrane gradually dries, cracks, and breaks up. In the process, fissures develop that impair epidermal barrier function, which can lead to percutaneous loss of water and permit the entry of microorganisms; fluid and electrolyte imbalances, skin infections, and sepsis may follow. In addition, circular bands of hardened skin can lead to vascular constriction and distal edema.
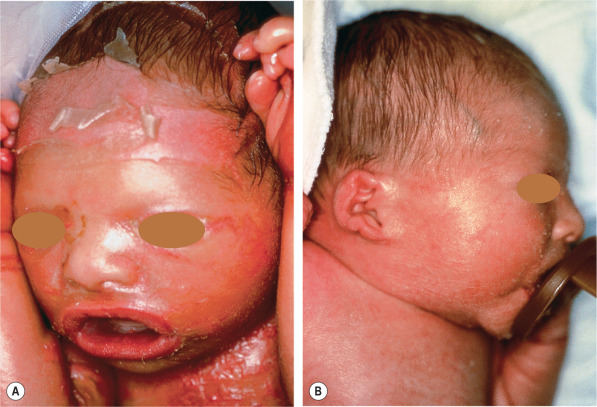
Within two to four weeks, the membrane peels off in sheets and a transition to the underlying disease phenotype takes place. Most commonly, congenital ichthyosiform erythroderma or lamellar ichthyosis ensues. Other ichthyoses may rarely present as a collodion baby ( Table 57.3 ). In self-improving collodion ichthyosis, desquamation of the collodion membrane leaves normal skin or variable clinical findings including xerosis, fine or focal scaling, palmar hyperlinearity, mild palmoplantar keratoderma, red cheeks, and hypohidrosis .
| DIFFERENTIAL DIAGNOSIS OF A COLLODION BABY | |
|---|---|
| Disorder | Frequency |
| Self-improving collodion ichthyosis | Always |
| Lamellar ichthyosis | Common |
| Congenital ichthyosiform erythroderma | Common |
| Trichothiodystrophy | Fairly common |
| Autosomal dominant lamellar ichthyosis | Rare |
| Autosomal dominant congenital ichthyosiform erythroderma | Rare |
| Sjögren–Larsson syndrome | Rare |
| Infantile Gaucher disease | Rare |
| Ichthyosis, intellectual disability, and spastic quadriplegia | Rare |
| Ectodermal dysplasias (EDs) * | Very rare |
| Neutral lipid storage disease | Very rare |
| Conradi–Hünermann–Happle syndrome ** | Very rare |
* E.g. hypohidrotic ED or ankyloblepharon–ED–clefting (AEC) syndrome.
** A collodion-like presentation is a feature of the allelic X-linked recessive multiple malformation syndrome affecting boys.
Pathology
Light and electron microscopic abnormalities of the collodion membrane are nonspecific and reveal mostly an excessively thickened, orthokeratotic stratum corneum. Therefore, it is usually preferable to perform multigene molecular testing or to defer a skin biopsy until transition to the underlying disease phenotype occurs. At that time, immunostaining can demonstrate the presence or absence of transglutaminase-1 protein in the upper epidermis . In situ assays of transglutaminase activity can also be performed on skin biopsy sections and allow rapid identification of patients with transglutaminase-1 deficiency, even as a collodion baby . However, interpretation of the transglutaminase activity assay requires experience, and this test is not widely available.
Differential Diagnosis
The phenotype of harlequin ichthyosis is much more severe and so striking that a clinical distinction between the two disorders is, in most circumstances, not difficult.
Treatment
Collodion babies are at risk for thermoinstability, hypernatremic dehydration, skin infections, and sepsis. It is therefore essential to keep them in a controlled environment and to carefully monitor for fluid and electrolyte imbalances. Collodion babies are also at risk for pneumonia secondary to restricted ventilation and aspiration of amniotic fluid containing scales. To facilitate gradual desquamation of the collodion membrane, it is generally recommended that infants be placed in a humidified incubator and the skin treated with wet compresses and light emollients to increase its elasticity and pliability. Manual removal of the collodion membrane and use of topical keratolytic agents are not advisable, because of the increased risk of infections and percutaneous absorption.
Lamellar Ichthyosis
▪ Non-erythrodermic autosomal recessive lamellar ichthyosis ▪ Ichthyosis congenita type 2
History
In 1966, Frost and Van Scott coined the term “lamellar ichthyosis” for all autosomal recessive forms of non-bullous congenital ichthyosis. Williams and Elias further separated this group into two distinct phenotypes, lamellar ichthyosis (LI) and (non-bullous) congenital ichthyosiform erythroderma (CIE), which now have been recognized as part of a continuum with intermediate phenotypes. In the current classification system, the LI–CIE spectrum as well as harlequin ichthyosis falls within the nonsyndromic autosomal recessive congenital ichthyosis (ARCI) category . However, a rare autosomal dominant form of LI with almost indistinguishable clinical features has also been described.
Epidemiology
LI occurs worldwide with an estimated prevalence of 1 in 200 000 to 1 in 300 000 live births, but it may be more common in certain regions such as Norway (1 in 90 000) or in inbred populations. LI is genetically heterogeneous and in most families is inherited as an autosomal recessive trait, although there are rare reports of autosomal dominant transmission.
Pathogenesis
Transglutaminase-1 deficiency due to deleterious mutations in both copies of TGM1 accounts for 35–55% of all patients with an ARCI on the LI–CIE spectrum and 65–90% of those with classic LI . LI–CIE spectrum patients with a collodion membrane at birth, plate-like scale, ectropion, and/or alopecia are approximately four times more likely to have TGM1 mutations than those without at least one of these features . Many patients of northern European descent share a German founder mutation that leads to alternative splicing of TGM1 mRNA .
Transglutaminase-1 catalyzes the calcium-dependent cross-linking of proteins through the formation of ε-(γ-glutamyl) lysine isopeptide bonds; it also creates ester bonds between proteins and ω-hydroxyceramides (see Fig. 56.3 ). This enzyme is expressed in the upper differentiated layers of the epidermis, where it facilitates the formation of the cornified cell envelope by cross-linking numerous structural proteins (e.g. involucrin, small proline-rich proteins, loricrin, keratins, desmosomal proteins) to one another as well as to the lipid envelope. Similar to human LI–CIE, transglutaminase-1 deficiency in transgenic mice severely perturbs development and maturation of the stratum corneum and postnatal adaptation of the skin to a dry environment.
Biallelic missense mutations in the ATP-binding cassette subfamily A member 12 gene ( ABCA12 ) have been reported primarily in LI patients of Northern African decent (Morocco, Mali, Algeria) . These mutations cluster within exons 28–32 which encode the first nucleotide-binding fold of this ABC transporter. The ABCA12 transporter is responsible for the energy-dependent transport of lipid substrates across membranes and is found in lamellar bodies. Of note, more deleterious mutations in ABCA12 cause harlequin ichthyosis (see below).
A mild form of LI can result from mutations in CYP4F22 , which encodes a cytochrome P450 enzyme involved in the ω-hydroxylation of ultra-long-chain fatty acids to form acylceramides; the latter are important to skin barrier function . Affected individuals have either a collodion membrane or erythroderma at birth, with later development of widespread whitish-gray scaling and palmoplantar hyperlinearity . Recently, mutations in the short chain dehydrogenase/reductase family 9C member 7 gene ( SDR9C7 ), which encodes a protein involved in vitamin A metabolism, were identified in patients with LI associated with recurrent dermatophyte infections . LI due to mutations in the sulfotransferase family 2B member 1 gene ( SULT2B1 ; encodes cholesterol sulfotransferase) presents with a collodion membrane at birth and then scaling that spares the central face and skin folds, reminiscent of steroid sulfatase deficiency . In addition, mutations in ALOX12B and ALOXE3 (encode lipoxygenases), NIPAL4 ( ICHTHYIN ), and LIPN (encodes lipase N) are occasionally associated with mild LI or intermediate LI/CIE phenotypes as well as the CIE end of the spectrum (see below).
Clinical Features
Classic LI is typically a severe disorder that is apparent at birth and persists throughout life, although milder variants also occur. Most affected neonates are encased in a collodion membrane (see previous section). Over the first weeks of life, large scales in a generalized distribution gradually replace the collodion membrane. These large, brown, plate-like scales form a mosaic or bark-like pattern and there is minimal to no associated erythroderma ( Fig. 57.11A, B ). In addition, the scales are centrally attached and have raised borders, often leading to superficial fissures. “Bathing suit ichthyosis”, which is caused by temperature-sensitive TGM1 mutations, affects primarily the trunk and scalp; it was first reported in South Africans and subsequently described in individuals from around the world .
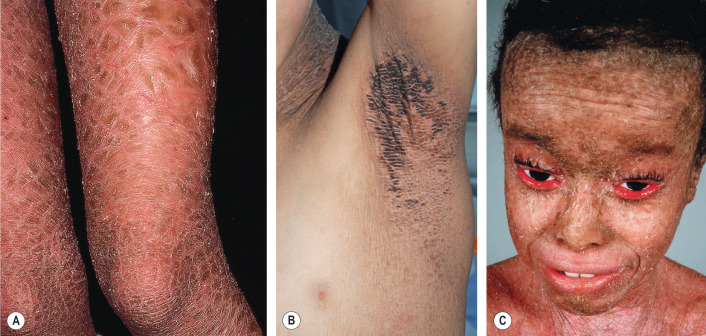
Tautness of facial skin commonly results in ectropion ( Fig. 57.11C ), eclabium, and hypoplasia of nasal and auricular cartilage. Severe ectropion may lead to madarosis, conjunctivitis, and incomplete lid closure with ensuing keratitis. Traction and compression exerted by the taut skin can cause scarring alopecia, especially at the periphery of the scalp. Hair shafts are normal but often encased by the thickened stratum corneum. The degree of palmoplantar keratoderma is variable and can range from accentuated skin markings to severe thickening with cracking and fissuring. Secondary nail dystrophy with thickened nail plates and ridging is not uncommon. Intraepidermal constriction of sweat ducts often results in severe heat intolerance, while accumulation of scale in the external ear canals can lead to occlusion, bacterial colonization, and recurrent infections.
Pathology
The histologic abnormalities are not diagnostic. Massive orthokeratotic hyperkeratosis covers an acanthotic epidermis, sometimes with psoriasiform or papillomatous hyperplasia. In contrast to CIE, the epidermal proliferation rate is normal or only slightly elevated. Elongated cholesterol clefts, variable numbers of translucent lipid droplets in the stratum corneum, and a thin or absent cornified cell envelope have been observed by electron microscopy, although these findings overlap with those of CIE .
Other diagnostic tests
In addition to measuring transglutaminase-1 activity in cultured keratinocytes, procedures to detect transglutaminase-1 deficiency in skin biopsy specimens include immunostaining with anti-transglutaminase-1 antibodies, in situ measurement of transglutaminase-1 activity (via covalent incorporation of biotinylated substrate peptides into skin cryostat sections) , and testing for the presence of cross-linked cell envelopes . However, these assays are not widely available for diagnostic purposes. Genetic testing to detect pathogenic mutations in TGM1 , ABCA12 , CYP4F22 , ALOX12B, ALOXE3, NIPAL4 (ICHTHYIN), SDR9C7, and LIPN is commercially available, including multigene panels . Molecular prenatal diagnosis can be performed in families with known mutations from chorionic villus sampling (CVS) or amniocentesis material obtained at an early gestational age ; this has largely replaced light and electron microscopic examination of fetal skin.
Differential Diagnosis
In the neonatal period, there is considerable clinical overlap with other congenital ichthyoses that present with a collodion membrane, especially CIE and self-improving collodion ichthyosis (see Tables 57.2 and 57.3 ). Later in life, the large, dark, plate-like scales, ectropion, and no discernible erythroderma of LI can be readily distinguished from most other ichthyoses. Although classic CIE is differentiated by marked erythroderma and small white scales, some patients exhibit intermediate LI/CIE phenotypes with variable degrees of erythroderma as well as quality and size of scales.
Treatment
Neonatal care is detailed in the collodion baby section. Severe disease often necessitates systemic therapy with oral retinoids from early childhood. Acitretin can be very effective in alleviating hyperkeratosis and scaling. Treatment is usually initiated at a low dose and then titrated to the minimal effective dose, which is dictated by the course and severity of the disease . The therapeutic benefits may also include an improvement of the ectropion, thus avoiding eye complications and reconstructive eyelid surgery. However, the benefits of long-term systemic retinoid therapy must be weighed against the potential toxicities. In a randomized controlled study of the retinoic acid metabolism blocking agent liarozole (not available in the US) in 64 patients with moderate-to-severe LI, a 41–50% response rate was observed in patients receiving liarozole, compared to 11% with placebo, with borderline statistical significance (p=0.056) ; in a previous study, liarozole had similar efficacy to acitretin but a more favorable tolerability profile .
Topical management should take into consideration the severely impaired desquamation and barrier function of the skin. The use of keratolytics is often limited due to skin irritation and an increased risk of systemic absorption, especially in children; systemic absorption of topical tacrolimus can also occur. Topical vitamin D 3 derivatives, tazarotene , and formulations containing lactic acid and propylene glycol in a lipophilic cream base have been effective. Heat intolerance can be ameliorated by frequent moistening of the skin with water or the use of air conditioning and humidifiers. Severe ectropion requires longitudinal ophthalmologic evaluation; surgical repair is sometimes necessary to prevent irreversible corneal damage.
LI and other severe forms of ARCI are often disfiguring, which may hinder the psychosocial development of affected children and adolescents. Families and patients need support in dealing with these issues, and patient advocacy groups such as FIRST ( www.firstskinfoundation.org ) are excellent resources.
Congenital Ichthyosiform Erythroderma
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


