Abstract
Hypertrichosis is defined as an excessive growth of hairs anywhere on the body. They are often terminal pigmented hairs but can be vellus or lanugo hairs. Hypertrichosis is frequently confused with hirsutism, but the latter term should be reserved for girls and women with excessive growth of terminal hairs in androgen-dependent sites. Hirsutism can result from hyperandrogenemia or increased end-organ sensitivity to androgens.
In addition to the type of hair, hypertrichosis is subdivided into congenital versus acquired as well as generalized versus localized. Acquired hypertrichosis lanuginosa can represent a paraneoplastic phenomenon while localized forms of hypertrichosis may be associated with trauma as well as underlying tumors or abnormalities of soft tissue and bone. Hypertrichosis is also a side effect of a number of medications. In addition to discontinuing incriminated drugs, treatment options include bleaching, physical and chemical depilatories, epilation, electrolysis, intense pulsed light therapy, and laser hair removal.
The evaluation of hirsutism involves measurement of serum androgen levels. In women, the ovaries and adrenal glands are the major sources of circulating androgens. If minimal or no hormonal abnormalities are detected, the hirsutism is categorized as constitutional or familial and presumed to be due to increased end-organ sensitivity. Depending on its etiology, treatment of hirsutism includes topical medications such as eflornithine, oral contraceptives, antiandrogens, glucocorticoids, gonadotropin releasing hormone agonists, and/or insulin-lowering agents in addition to the physical measures outlined for hypertrichosis.
Keywords
hypertrichosis, generalized congenital hypertrichosis, generalized acquired hypertrichosis, hypertrichosis lanuginosa, prepubertal hypertrichosis, drug-induced hypertrichosis, Becker melanosis, nevoid hypertrichosis, hirsutism, constitutional hirsutism, hyperandrogenemia, hyperandrogenism, SAHA syndrome, HAIR-AN syndrome, antiandrogens, glucocorticoids, gonadotropin-releasing hormone agonists
- ▪
Hypertrichosis refers to excessive growth of hair anywhere on the body, whereas hirsutism represents excessive hair growth within androgen-dependent sites in girls and women
- ▪
Hypertrichosis may be generalized or localized, with etiologies ranging from genodermatoses to underlying hamartomas to repeated trauma
- ▪
Hirsutism is related to hormonal factors, in particular an increase in circulating androgen levels and/or enhanced sensitivity of hair follicles to androgens
- ▪
In women, the major sources of androgens are the adrenal glands and the ovaries; dysfunction of these organs must be considered when a patient presents with hirsutism
- ▪
Depending on its etiology, treatment of hirsutism includes antiandrogens, glucocorticoids, insulin-lowering agents, and/or contraceptives, combined with topical medications and physical measures (e.g. laser hair removal)
Hypertrichosis
Introduction
Hypertrichosis describes the growth of excessive hair anywhere on the body. The term is frequently confused with hirsutism, which should only be applied to women with excessive growth of terminal hairs in androgen-dependent sites (i.e. a “male pattern”) due to hyperandrogenemia or increased end-organ sensitivity to androgens . Hypertrichosis can be classified based on its distribution (generalized versus localized), the age of onset (congenital or programmed from birth versus acquired), and the type of hair (lanugo or vellus versus terminal).
Clinical Features
Generalized hypertrichosis
In generalized hypertrichosis, there is lanugo hair, excess vellus hair, or terminal hair over much of the cutaneous surface, including an acquired transformation of terminal hair into lanugo hair ( Fig. 70.1 ) . Lanugo is the non-pigmented, non-medullated, fine hair that covers the fetus and can grow up to several centimeters in length; it is normally shed in utero or during the first few weeks of life and replaced by vellus hair on the body and terminal hair on the scalp. Age of onset can vary from infancy to prepubertal to adulthood.
Congenital generalized hypertrichosis
A number of distinctive but rare genetic syndromes are associated with congenital generalized hypertrichosis ( Table 70.1 ). While the majority of these disorders have extracutaneous manifestations such as gingival hyperplasia or facial dysmorphism, some have primarily hypertrichosis, including universal hypertrichosis. However, this latter entity is sometimes viewed as constitutional, i.e. simply exaggerated normal hairiness that may be familial. Genetic abnormalities associated with congenital generalized hypertrichosis lead to dysfunction of several proteins, ranging from those known to be involved in hair follicle development to membrane transporters (see Table 70.1 ).
| HEREDITARY DISORDERS CHARACTERIZED BY CONGENITAL GENERALIZED HYPERTRICHOSIS | ||
|---|---|---|
| Disorder | Inheritance (locus; genetic basis) | Other key features |
| Increased hair is the major feature | ||
| Congenital hypertrichosis lanuginosa | AD |
|
| Universal hypertrichosis | AD |
|
| Ambras syndrome (hypertrichosis universalis congenita, Ambras type; HTC1) | AD (8q22–q24 breakpoints; position effect down-regulates TRPS1 expression) |
|
| Generalized hypertrichosis with extracutaneous features | ||
| X-linked hypertrichosis (congenital generalized hypertrichosis; HTC2) | XLD (Xq27.1; palindrome-mediated interchromosomal insertion; position effect down-regulates FGF13 expression) |
|
| Congenital generalized hypertrichosis with or without gingival hyperplasia (HCT3) | AD or AR (17q24.2–q24.3 microdeletion or microduplication [AD]; position effect down-regulates SOX9 expression; also ABCA5 mutations [AR]) |
|
| Cantú syndrome (hypertrichotic osteochondrodysplasia) | AD (12p21.1; ABCC9 mutation) |
|
| Zimmermann–Laband syndrome 1 & 2 |
|
|
| Coffin–Siris syndrome 1–5 ** |
|
|
| Schinzel–Giedion midface retraction syndrome | AD (18q12.3; SETBP1 mutation) |
|
| Gorlin–Chaudry– Moss syndrome | AR vs XLD |
|
| Adducted thumbs syndrome | AR |
|
| Barber–Say syndrome | AD (2q37.3; TWIST2 mutation) |
|
| Amaurosis congenita, cone–rod type, with congenital hypertrichosis ‡ | AR |
|
| CAHMR syndrome ‡ | AR |
|
* This pattern of distribution is also observed in prepubertal hypertrichosis (see text).
** Protein products of mutated genes are subunits of the SWI/SNF complex.
The possibility of intrauterine exposure to medications (e.g. minoxidil) also needs to be considered in infants with congenital generalized hypertrichosis. The differential diagnosis also includes inherited disorders in which hypertrichosis can involve multiple sites and may appear early in life ( Table 70.2 ).
| HEREDITARY DISEASES AND CONGENITAL SYNDROMES ASSOCIATED WITH REGIONAL HYPERTRICHOSIS | ||
|---|---|---|
| Disorder | Sites of hypertrichosis | Other key features |
| Dysmorphic syndromes | ||
| Cornelia de Lange syndrome 1–5 |
|
|
| Rubinstein–Taybi syndrome 1 & 2 |
|
|
| Disorders with primary cutaneous features | ||
| Porphyrias |
|
|
| Lipodystrophy syndromes (e.g. Berardinelli–Seip syndrome, leprechaunism) |
|
|
| Erythrokeratodermia variabilis |
|
|
| Dystrophic epidermolysis bullosa * |
|
|
| Ichthyosis bullosa of Siemens * |
|
|
| Metabolic disorders | ||
| Mucopolysaccharidoses † |
|
|
| Congenital hypothyroidism |
|
|
| Mitochondrial disorders | ||
| Leigh syndrome due to SURF1 mutations |
|
|
| MELAS syndrome |
|
|
| Intrauterine exposures ‡ | ||
| Fetal hydantoin syndrome |
|
|
| Fetal alcohol syndrome |
|
|
| Morpheaform or sclerodermoid disorders | ||
| MONA (multicentric osteolysis, nodulosis, and arthropathy; previously known as Winchester syndrome) |
|
|
| H syndrome § , |
|
|
| Stiff skin syndrome |
|
|
| Linear melorheostosis (isolated melorheostosis) ¶ |
|
|
* Hypertrichosis is an uncommon feature.
† Hypertrichosis may also be observed in other lysosomal storage diseases, e.g. Krabbe disease and GM1-gangliosidosis, as well as sialuria.
‡ Also minoxidil and diazoxide.
§ Allelic with pigmented hypertrichosis with insulin-dependent diabetes mellitus (PHID) syndrome, sinus histiocytosis with massive lymphadenopathy (SHML), and Faisalabad histiocytosis.
¶ Hypertrichosis has also been described overlying linear scleroderma in the absence of melorheostosis.
Prepubertal hypertrichosis
Prepubertal hypertrichosis is a relatively common finding in otherwise healthy infants and children, most often occurring in individuals of Mediterranean or South Asian descent. Pigmented hair is present in a widespread, diffuse distribution and becomes more obvious during childhood. There is involvement of the face (especially the forehead, temples, and preauricular area), proximal extremities, and back; hairs in the latter location assume an “inverted fir tree” pattern. Bushy eyebrows and a low anterior hairline represent additional features.
The facial distribution pattern of prepubertal hypertrichosis can overlap with that of familial hirsutism (see below), and there may be a family history of excessive hairiness. Mildly elevated levels of total and free testosterone have been observed in a subset of girls with prepubertal hypertrichosis, while others have a normal androgen profile . These findings suggest multiple etiologies for this clinical pattern of hypertrichosis, including androgen excess as well as an increased constitutional propensity for hair growth.
Acquired generalized hypertrichosis
Acquired generalized hypertrichosis is most often related to drug ingestion ( Table 70.3 ). Drug-induced hypertrichosis is characterized by slow growth of terminal hair of medium thickness. The findings are most evident on the forehead, temples, flexor aspects of the extremities, and trunk. Drug-related hypertrichosis is usually reversible, and differs in distribution from drug-induced hirsutism.
| HYPERTRICHOSIS DUE TO DRUGS | |
|---|---|
| Antibiotics | |
| |
| Anti-inflammatory drugs | |
|
|
| Vasodilators | |
|
|
| Diuretics | |
| |
| Anticonvulsants | |
| |
| Immunosuppressives | |
|
|
| Psoralens | |
|
|
| Antiseptic agents | |
| |
| Chelators | |
| |
| Other | |
|
|
Acquired generalized hypertrichosis can also represent a sign or consequence of a variety of systemic conditions, including disorders of the CNS (e.g. traumatic brain injuries), juvenile hypothyroidism, juvenile dermatomyositis, acromegaly (especially of the lower face), malnutrition (including anorexia nervosa), POEMS syndrome, and advanced HIV infection.
Acquired hypertrichosis lanuginosa
This is considered to be a paraneoplastic phenomenon as it is associated with internal malignancies, most often of the lung, colon, or breast. Occasionally, acquired hypertrichosis lanuginosa may precede the diagnosis of the neoplasm. In addition, it may be associated with other paraneoplastic dermatoses, such as acanthosis nigricans, palmoplantar keratoderma, the sign of Leser–Trélat, and acquired ichthyosis (see Ch. 53 ). The lanugo hair appears over the entire body within a short period of time, although in mild forms it may be localized to the face, leading to a “simian” appearance. Lanugo hair may even develop in areas of androgenetic alopecia.
Localized hypertrichosis
Most cases of localized hypertrichosis involve a switch from vellus to terminal hair in sites that do not usually bear terminal hair. Localized hypertrichosis can develop as a component of a hamartoma, as an isolated congenital lesion, as a manifestation of a systemic disease (inherited or acquired), or as a consequence of cutaneous trauma or inflammation.
Congenital localized hypertrichosis
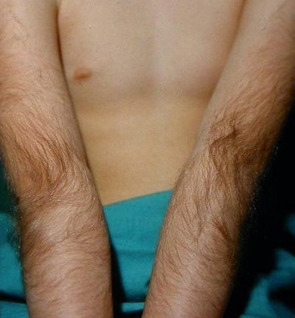
Hamartomas, including those with delayed clinical presentation, and congenital abnormalities characterized by hypertrichosis of a specific anatomic site ( Fig. 70.2 ; Tables 70.4 & 70.5 ) are included in this category.
| HEREDITARY HYPERTRICHOSIS AFFECTING SPECIFIC ANATOMIC SITES | |||
|---|---|---|---|
| Condition | Inheritance | Onset | Other features |
| Hypertrichosis cubiti (hairy elbow syndrome; Fig. 70.2 ) | AD | Birth to early childhood |
|
| Hairy palms and soles | AD | Birth | |
| Hypertrichosis of the auricle | AD † | Childhood or adolescence |
|
| Hypertrichosis of the eyebrows | ? | Adolescence | |
| Trichomegaly of the eyelashes | AR | Childhood | |
| Hypertrichosis of the nasal tip | ? | Adolescence |
|
| Anterior cervical hypertrichosis ( Fig. 70.5 ) | AD ‡ | Birth to early childhood | |
| Posterior cervical hypertrichosis | AD | Birth |
|
| Polythelia, including the hairy variant § , | AD ¶ | Adolescence |
|
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


