Abstract
Heritable disorders of connective tissue comprise a phenotypically diverse group of conditions that often have multisystem involvement. These disorders can result from mutations in genes encoding structural elements of the extracellular matrix (ECM; e.g. collagens, components of elastic fibers) as well as enzymes and other proteins that have effects on the ECM. Recognition of characteristic skin findings is often the key to establishing the diagnosis and identifying associated internal manifestations. This chapter provides a detailed review of Ehlers–Danlos syndrome, pseudoxanthoma elasticum, and cutis laxa and briefly discusses other genetic disorders of the ECM.
Keywords
Ehlers–Danlos syndrome, pseudoxanthoma elasticum, cutis laxa, Marfan syndrome, homocystinuria, Buschke–Ollendorff syndrome, collagen, elastic fibers, extracellular matrix
- ■
Heritable connective tissue disorders with skin involvement comprise a phenotypically diverse group of conditions
- ■
Recognition of characteristic skin findings is often critical to establishing the diagnosis and identifying associated internal involvement, which may include life-threatening cardiovascular disease as in vascular Ehlers–Danlos syndrome and pseudoxanthoma elasticum
- ■
Heritable connective tissue disorders can result from mutations in genes encoding structural extracellular matrix (ECM) proteins (e.g. collagens, components of elastic fibers), enzymes that modify these structural proteins, and other proteins with effects on the ECM (e.g. via transport or regulatory functions)
Introduction
Heritable connective tissue diseases manifest with a broad range of phenotypic abnormalities, and variable degrees of cutaneous and extracutaneous involvement. At one end of the spectrum, the clinical findings are mild and may be limited to the skin, while at the other end of the spectrum, the cutaneous manifestations are part of multi-organ pathology causing considerable morbidity and even mortality . There have been significant advances in understanding the molecular bases of heritable disorders of connective tissue. This has led to improved classification and prognostication, with better recognition of phenotypic spectrums and relationships among disorders.
Connective tissue is critical for organogenesis during embryonic development and homeostatic maintenance of tissues, with complex protein assembly and cell-matrix interactions. The principal fibrillar components of the extracellular matrix (ECM) of the skin are networks consisting of collagen and elastic fibers (see Ch. 95 ). The study of heritable disorders of connective tissue has demonstrated the considerable impact of disruption of various individual components of the ECM and related proteins.
Classic, “primary” heritable disorders of connective tissue caused by mutations in the genes encoding collagen and structural components of elastic fibers include several forms of Ehlers–Danlos syndrome (EDS) and cutis laxa, respectively. In addition to defects in the genes that encode these structural proteins and enzymes that modify them, seemingly unrelated pathologic processes can perturb the ECM. Examples of the latter, which can be considered as “secondary” connective tissue disorders, include pseudoxanthoma elasticum (PXE) and homocystinuria.
This chapter provides a detailed review of EDS, PXE, and cutis laxa. In addition, Marfan syndrome, homocystinuria, osteogenesis imperfecta, and Buschke–Ollendorff syndrome are presented in Table 97.1 . These and other genetic diseases that affect the ECM of the skin are also summarized in Table 95.5 .
| ADDITIONAL HERITABLE DISORDERS OF CONNECTIVE TISSUE WITH CUTANEOUS FINDINGS | ||||
|---|---|---|---|---|
| Disease | Cutaneous findings | Extracutaneous manifestations | Inheritance | Associated gene/protein product |
| Marfan syndrome | Striae, elastosis perforans serpiginosa, decreased subcutaneous fat on extremities | Skeletal abnormalities (e.g. disproportionately long limbs, arachnodactyly, scoliosis), lens subluxations (upward), dilation/dissection of ascending aorta * | AD | FBN1 /fibrillin 1 defects (the major component of microfibrils) † |
| Homocystinuria | Malar flush, livedo reticularis, diffuse pigmentary dilution, tissue paper-like scars | Marfanoid habitus, lens subluxations (downward), venous and arterial thrombosis ‡ , mental retardation | AR | CBS /cystathionine synthase enzyme deficiency (resulting in increased plasma homocysteine § ); rarely, methylenetetrahydrofolate reductase, 5-methyltetrahydrofolate-homocysteine methyltransferase, or 5-methyltetrahydrofolate-homocysteine methyltransferase reductase deficiency (enzymes that convert homocysteine back to methionine) |
| Osteogenesis imperfecta | Thin skin | Bone fragility, blue sclerae, dentinogenesis imperfecta (in a subset of patients) | AD | COL1A1 , COL1A2 /type I collagen defects |
| Buschke–Ollendorff syndrome | Connective tissue nevi (dermatofibrosis lenticularis disseminata); occasionally cutaneous sclerosis associated with melorheostosis | Osteopoikilosis (“spotted” bones); occasionally melorheostosis (sclerotic bone resembling “dripping candle wax” on radiographs; reflects a postzygotic “second hit” in the LEMD3 gene); rarely otosclerosis | AD | LEMD3 /LEM domain-containing 3, an antagonist of bone morphogenic protein (BMP) and TGF-β signaling |
* In addition to traditional treatment with β-blockers, angiotensin II receptor blockers (e.g. losartan), which antagonize pathogenic transforming growth factor (TGF)-β signaling, can prevent aortic root dilation in patients with Marfan syndrome; administration of doxycycline, which inhibits metalloproteinases-2 and -9, may also be of benefit.
† Mutations in the genes encoding TGF-β receptors 1 and 2 can lead to a phenotype that mimics Marfan syndrome.
‡ Methionine-restricted, cysteine-supplemented diet plus pyridoxine recommended.
§ Elevated plasma homocystine and homocysteine can be detected in patients with homocystinuria, but only homocysteine is elevated in hyperhomocysteinemia.
Ehlers–Danlos Syndrome
▪ Cutis hyperelastica
- ■
Clinically and genetically heterogeneous group of connective tissue disorders caused by defective function of various collagens, ECM-processing enzymes, and the collagen-associated protein tenascin-X
- ■
Autosomal dominant and autosomal recessive inheritance patterns
- ■
Typical cutaneous features include hyperextensible and fragile skin, easy bruising, and poor wound healing
- ■
Variable extracutaneous features include joint hypermobility, scoliosis, ocular changes, and, particularly in the vascular type, a significant risk of arterial, intestinal, or uterine rupture
History
Patients with features of Ehlers–Danlos syndrome (EDS) were described as early as the mid-1600s. The first comprehensive report of this condition was published in 1892 by Tschernogobow . Drs Ehlers and Danlos, Danish and French dermatologists, respectively, contributed to a comprehensive clinical description in the early 1900s, and this constellation of findings became known as the Ehlers–Danlos syndrome in 1936. The genetic nature of EDS was noted in 1949, and soon thereafter it was suggested that the phenotype resulted from abnormal collagen. The genetic heterogeneity of EDS became evident in the 1960s, and molecular defects in collagen synthesis were first described in 1972.
In the past, EDS was divided into 11 subtypes (numbered I–XI) based primarily upon clinical features. In 1997, a consensus conference was held and a revised nosology was proposed . Currently, the molecular basis of most major forms of EDS is known and a new international classification was proposed in 2017, recognizing 13 subtypes ( Table 97.2 ).
| CLASSIFICATION OF EHLERS–DANLOS SYNDROME | ||||
|---|---|---|---|---|
| EDS type | Historical classification † | Clinical features | Inheritance | Mutated gene/protein |
| Classic * | I, II | Hyperextensible skin, joint hypermobility, atrophic scars, easy bruising | AD | COL5A1, COL5A2 /α 1 – and α 2 -chains of type V collagen Rarely, COL1A1 /α 1 -chain of type I collagen (associated with vascular fragility) |
| Classic-like EDS | Similar to mild classic type but normal scarring | AR | TNXB, tenascin-X | |
| Cardiac valvular | Cardiac valve defects, otherwise similar to the classic type | AR | COL1A2 /α 2 -chain of type I collagen | |
| Hypermobile * | III | Joint hypermobility, pain and dislocations; less pronounced skin changes; variable autonomic dysfunction | AD | Unknown but ~10% of affected individuals, predominantly female, show haploinsufficiency of tenascin-X ( TNXB ) |
| Vascular * | IV | Thin, translucent skin; arterial, gastrointestinal, and uterine rupture; excessive bruising, small joint hypermobility, characteristic facies | AD | COL3A1 /α 1 -chain of type III collagen |
| Kyphoscoliotic * | VIA | Hyperextensible skin, easy bruising, joint hypermobility, kyphoscoliosis, congenital hypotonia, blue sclerae, marfanoid habitus PLOD1 : atrophic scarring, ocular fragility FKBP14 : follicular hyperkeratosis, myopathy, impaired hearing | AR | PLOD1 /lysyl hydroxylase § FKBP14 /FK506-binding protein 14 |
| Musculocontractural | VIB | Hyperextensible and fragile skin, wrinkled palms, joint contractures and hypermobility, scoliosis, ocular fragility, craniofacial abnormalities | AR | CHST14 /dermatan-4-sulfotransferase 1 (allelic with adducted thumb–clubfoot syndrome) DSE/ dermatan sulfate epimerase |
| Arthrochalasia * | VIIA, VIIB | Hyperextensible and fragile skin, severe joint hypermobility with congenital bilateral hip dislocation, scoliosis | AD | COL1A1 , COL1A2 /α 1 – and α 2 -chains of type I collagen |
| Dermatosparaxis * | VIIC | Severe skin fragility, sagging and redundant skin that is soft and doughy, easy bruising, hernias, distinctive facies | AR | ADAMTS2 /procollagen I N-peptidase |
| Periodontal | VIII | Mildly hyperextensible skin, excessive bruising, small joint hypermobility, early-onset periodontitis with gingival recession; increased infections, marfanoid habitus | AD | C1R, C1S/ complement C1r, C1s |
| Spondylodysplastic (includes former progeroid and spondylocheirodysplasia types) | Soft/doughy, translucent, and/or hyperextensible skin, variable joint hypermobility, hypotonia, limb bowing, short stature, variable tapered fingers, osteopenia, developmental delay B4GALT6/7 : progeroid facies, joint contractures SLC39A13 : wrinkled palms, platyspondyly, protuberant eyes | AR | B4GALT7 /galactosyl transferase-1, B3GALT6/ galactosyltransferase-II SLC39A13 /zinc transporter ZIP13 | |
| Brittle cornea syndrome | Soft, translucent skin with variable hyperextensibility and fragility, acral wrinkling, hypermobility of small joints, ocular fragility, keratoconus, blue sclerae, impaired hearing, scoliosis | AR | ZNF469/ zinc finger protein 469 PRDM5/ PR domain-containing protein 5 | |
| Myopathic | Soft/doughy skin with atrophic scarring, proximal joint contractures and distal hypermobility, congenital myopathy with hypotonia that improves with age | AD or AR | COL12A1 /α 1 -chain of type XII collagen | |
* Six main types recognized by the 1997 Villefranche consensus classification .
† Replaced by the 1997 Villefranche consensus classification and now the 2017 international classification, which recognizes 13 subtypes .
§ Analysis of the urine for an increased deoxypyridinoline : pyridinoline ratio can be used as a screening test for kyphoscoliosis-type EDS.
Epidemiology
Features suggestive of EDS (e.g. loose-jointedness) are present in a relatively large number of individuals in the general population, especially in certain African groups. However, more stringent diagnostic criteria that include findings in the skin and joints, at a minimum, estimate the incidence of EDS to be ~1 in 5000.
Pathogenesis
EDS is a genetically as well as clinically heterogeneous group of connective tissue disorders that most often result from disruption of collagen fibrils ( Fig. 97.1 ). The collagen family includes 28 distinct proteins that differ in their tissue distributions and functions (see Ch. 95 ). Each collagen consists of three α-chain polypeptides, many of which are synthesized as precursor pro-α-chains; a trimeric collagen molecule may be either a homotrimer or heterotrimer consisting of the same versus two or three genetically different kinds of polypeptides, respectively. There are 45 distinct α-chains, each encoded by a different gene .

The pathogenesis of EDS can involve mutations in genes for collagen-processing enzymes as well as those encoding collagen α-chains. The latter result in either dominant-negative effects, e.g. disruption of trimers and fibrils due to incorporation of mutant collagen α-chains, or haploinsufficiency. The precise phenotype depends upon the nature of the mutation(s) and the type of collagen or other protein that is affected.
Several forms of EDS are caused by dominant-negative mutations in the genes encoding α-chains of type I ( COL1A1, COL1A2 ), type III ( COL3A1 ), and type V ( COL5A1, COL5A2 ) collagens. Characteristic examples include splice site mutations that can result in an in-frame deletion of a single exon and missense mutations that lead to a glycine substitution in the collagenous domain. The resultant “mutant” polypeptides are incorporated into the polymeric collagen molecules and disrupt the formation, secretion, and/or thermal stability of triple helices. As a consequence, the collagen fibrils are often reduced in amount as well as structurally abnormal.
Haploinsufficiency is caused by a heterozygous loss-of-function mutation, usually associated with a premature termination codon. These types of mutations often result in unstable mRNA that undergoes accelerated decay, leading to an absence of the corresponding polypeptide. Haploinsufficiency can also be caused by a missense mutation encoding a polypeptide that cannot be incorporated into collagen triple helices. Haploinsufficiency occurs when the synthesis of only 50% of the normal amount of the collagen is not enough to form fully functional fibrils, thus resulting in a disease phenotype.
Vascular EDS can result from either dominant-negative mutations in COL3A1 or haploinsufficiency of type III collagen, with the latter associated with a delayed onset of arterial complications and prolonged survival . Collagen III is an important component of arterial and intestinal walls and a less abundant component of dermal connective tissue. However, a reduced amount of type III collagen leads to small or variably sized dermal collagen fibrils and thinning of the dermis, emphasizing the role of type III collagen in fibrillogenesis. A decreased amount of collagen III in the dermis has also been detected in some patients with the periodontal and kyphoscoliotic types of EDS which, unlike vascular EDS, are not caused by COL3A1 mutations.
Classic EDS can be caused by dominant-negative mutations in COL5A1 and COL5A2 or (in 30–50% of cases) haploinsufficiency of type V collagen ( COL5A1 ). The role of type V collagen in collagen fibrillogenesis is to limit fibril diameter, explaining the ~25% increase in fibril diameter that is observed by electron microscopy in affected individuals.
Arthrochalasia EDS results from heterozygous COL1A1 and COL1A2 mutations that specifically affect the amino-terminal cleavage site of procollagen I N-peptidase and thereby disrupt processing of type I collagen pro-α-chains. Biallelic loss-of-function mutations in ADAMTS2 which encodes this procollagen I N-peptidase are responsible for dermatosparaxis EDS. The latter disorder is characterized by irregular and thin collagen fibrils containing type I collagen with an intact N-propeptide, termed pN-collagen. Kyphoscoliotic EDS is caused by biallelic mutations in PLOD1 , which encodes another collagen-processing enzyme, lysyl hydroxylase; this leads to reduced hydroxylysine content in collagen polypeptides and altered cross-linking of collagen fibrils.
Complete tenascin-X deficiency via biallelic null mutations underlies an autosomal recessive form of EDS with features similar to those of classic EDS . Tenascin-X is an ECM protein that is thought to reinforce associations among collagen fibrils and may also regulate the structure and stability of elastic fibers. EDS can also result from defective glycosaminoglycan processing due to mutations in genes encoding galactosyltransferases ( B4GALT6, B3GALT7 ), dermatan-4-sulfotransferase 1 ( CHST14 ), and dermatan sulfate epimerase ( DSE ) (see Ch. 95 ). Other EDS types occur through diverse mechanisms including disruption of intracellular zinc homeostasis (solute carrier family 39, member 13 [ SLC39A13 ]), protein folding in the endoplasmic reticulum (FK506-binding protein 14 [ FKBP14 ]), and the classical complement pathway ( C1R , C1S ).
Clinical Features
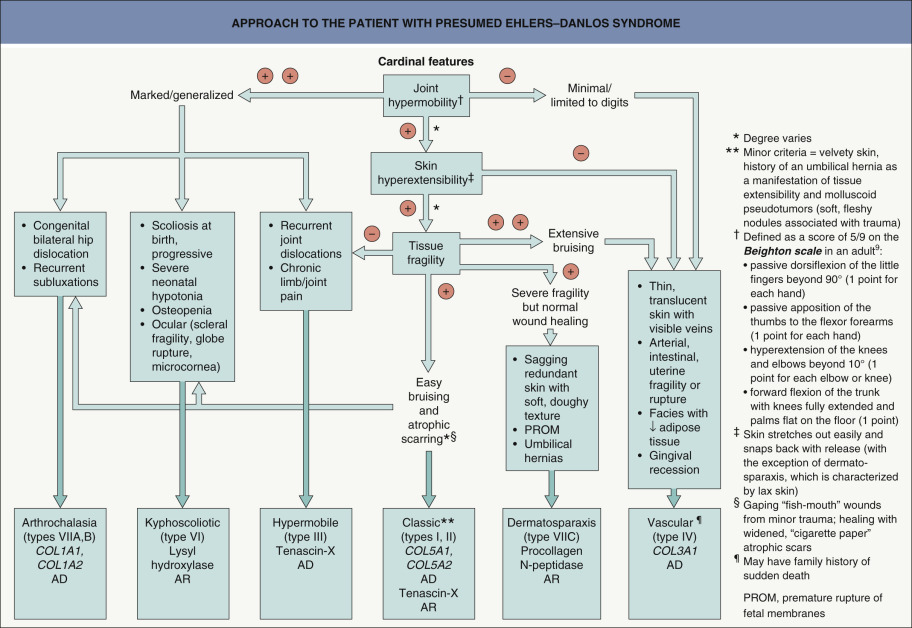
The general features of EDS include hyperextensible fragile skin and joint hypermobility ( Fig. 97.2A, B ). The Beighton scale can be utilized to assess the latter , and different subtypes of EDS have additional manifestations that allow their classification into the six major categories defined by the 1997 consensus conference ( Fig. 97.3 ). Table 97.2 outlines the other recognized variants of EDS.
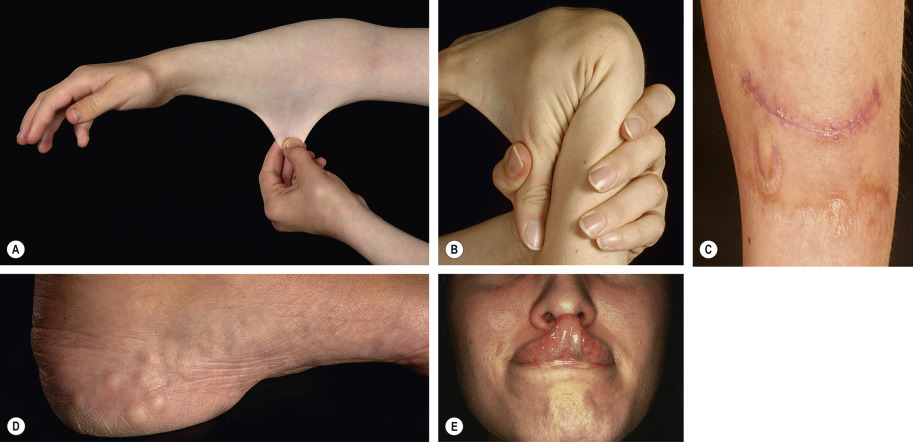
Typical skin findings are present, to a varying degree, in all subtypes of EDS . Skin hyperextensibility can be assessed on the volar surface of the distal forearm or wrist, where the upper limit of normal extensibility is 1–1.5 cm. The skin also tends to be fragile, splitting readily due to trauma, particularly on the knees, elbows, shins, and forehead. These wounds present with a gaping, “fish-mouth” appearance; healing may be delayed and dehiscence is common. The resulting scars are typically atrophic and widen over time ( Fig. 97.2C ), although some affected individuals develop hypertrophic scars. Sites of repeated trauma develop a variety of secondary lesions such as molluscoid pseudotumors, which are fleshy nodules of redundant skin and fibrous tissue that favor the knees and elbows. Multiple small, hard, mobile nodules over bony prominences such as the ulnar and tibial borders are also a frequent finding. These subcutaneous spheroids are thought to represent calcified fat globules. Easy bruising is commonly observed, reflecting the fragility of small blood vessels. Dermatologic disorders that are associated with, but not specific to, EDS include elastosis perforans serpiginosa and piezogenic papules ( Fig. 97.2D ).
Classic EDS is inherited in an autosomal dominant fashion and presents with hypermobile joints as well as hyperextensible fragile skin that is prone to bruising. Scars are typically widened and atrophic, often with a “cigarette paper” appearance. The skin also tends to be soft, with a slightly doughy or velvety texture. At least half of affected individuals can touch the tip of their nose with their tongue (Gorlin sign), compared to 10% of the general population ( Fig. 97.2E ). Although classic EDS may be associated with mild aortic root dilation and mitral valve prolapse, each in ~5% of patients, these findings are rarely of clinical significance . Other frequent musculoskeletal manifestations include pes planus, hypotonia, and temporomandibular joint dysfunction.
Patients with tenascin-X deficiency , which has autosomal recessive inheritance, have a phenotype similar to that of classic EDS but with relatively normal scarring. This variant is occasionally associated with congenital adrenal hyperplasia due to deletion of the contiguous gene encoding 21-hydroxylase.
Hypermobile EDS is the most common variant and has autosomal dominant inheritance. The most prominent clinical feature is generalized joint hypermobility, typically leading to recurrent joint dislocations that can cause chronic pain and eventual osteoarthritic changes. Easy bruising is common, and the skin tends to be soft but only mildly hyperextensible. Patients may have autonomic dysfunction presenting with orthostatic intolerance and gastrointestinal symptoms .
Vascular EDS is an autosomal dominant disorder characterized by thin translucent skin with extensive bruising and a high risk of arterial, intestinal, and uterine ruptures . Spontaneous arterial rupture has the highest incidence in the third and fourth decades of life but may occur earlier, and median survival is ~50 years . Affected individuals often have a characteristic facial appearance, with a “pinched” nose, thin lips, micrognathia, hollow cheeks, and prominent “staring” eyes due to decreased periorbital adipose tissue. Small earlobes and fine scalp hair are additional features.
Kyphoscoliotic EDS is an autosomal recessive disorder that features skin hyperextensibility, generalized joint hypermobility, severe hypotonia at birth, delayed gross motor development, progressive congenital scoliosis, and scleral fragility that can lead to rupture of the ocular globe. Additional findings may include atrophic scars, easy bruising, fragility of other tissues (rarely arterial rupture), a marfanoid habitus, microcornea, and osteopenia.
Arthrochalasia EDS is inherited in an autosomal dominant fashion. The major clinical features include severe generalized joint hypermobility with recurrent subluxations and congenital bilateral hip dislocation. Patients may also present with hyperextensible skin, atrophic scars, easy bruising, hypotonia, scoliosis, and mild osteopenia.
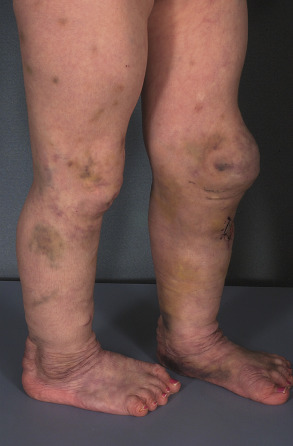
Dermatosparaxis EDS is a very rare autosomal recessive disorder that, unlike other forms of EDS, presents with skin that is sagging and redundant as well as extremely fragile. The skin has a highly characteristic soft, doughy consistency, and it tears and bruises easily ( Fig. 97.4 ). Additional findings include premature rupture of fetal membranes, large umbilical or inguinal hernias, delayed closure of the fontanelles, distinctive facies with puffy eyelids and micrognathia, blue sclerae, gingival hyperplasia, abnormal dentition, and short limbs.
Pathology and Laboratory Findings
Histopathologic examination of the skin is usually noncontributory. Nonspecific increases in elastic fibers have been reported, particularly in areas of repeated trauma or molluscoid pseudotumors. However, in vascular EDS the dermis is often reduced to ~25% of its normal thickness, and immunohistochemical staining may show intracellular accumulation of type III collagen.
Electron microscopy in classic EDS often shows collagen fibrils with variable diameters and occasional large “cauliflower” fibrils with an irregular contour. Ultrastructurally, arthrochalasia and dermatosparaxis types of EDS are characterized by angular and “hieroglyphic” appearances of collagen fibrils in cross-section, respectively.
Traditionally the diagnosis of EDS has been based upon clinical findings, but nowadays genetic testing is available for all the major forms of EDS except for the hypermobile type. Protein electrophoresis of collagens extracted from cultured dermal fibroblasts can be helpful in demonstrating specific collagen defects, particularly in the vascular, arthrochalasia, and dermatosparaxis types of EDS . Analysis of the urine for an increased deoxypyridinoline : pyridinoline ratio may be used as a screening test for kyphoscoliotic-type EDS, and tenascin-X deficiency can be demonstrated by an ELISA of a serum sample .
Differential Diagnosis
In addition to the major forms of EDS discussed above, other uncommon variants should also be considered (see Table 97.2 ). Several previously described rare subtypes of EDS, including type V (X-linked variant) and type X (fibronectin type), are currently excluded from the EDS nosology due to a lack of clear descriptions of their clinical features and unknown molecular bases. The previous EDS type IX, an X-linked recessive condition known as occipital horn syndrome, is now considered to represent a form of cutis laxa (see Table 97.6 ) rather than EDS. In contrast to cutis laxa, which is characterized by loose and sagging skin with decreased resilience, the skin of patients with EDS (except the dermatosparaxis type) retains its elasticity and recoil. Although some patients with EDS, in particular the kyphoscoliotic type, have a marfanoid body habitus, they do not typically exhibit other findings of Marfan syndrome ( Table 97.3 ), a distinct heritable connective tissue disorder due to mutations in the fibrillin 1 gene ( FBN1 ) (see Table 97.1 ).
| COMPARISON OF SELECTED CLINICAL FEATURES IN EHLERS–DANLOS SYNDROME AND MARFAN SYNDROME | ||
|---|---|---|
| Ehlers–Danlos syndrome | Marfan syndrome | |
| Molluscoid pseudotumors | ++ | − |
| Skin | Hyperextensibility, “fish-mouth” atrophic scars | Striae |
| Joint hypermobility | ++ (often severe) | + (mild–moderate) |
| Easy bruising | ++ | − |
| Pectus excavatum | − | ++ |
| Scoliosis | + | ++ |
| High-arched palate | − | ++ |
| Ectopia lentis | + | ++ |
| Myopia | + | ++ |
| Mitral valve abnormalities | + | ++ |
| Stature | Variable | Tall |
The phenotype of Loeys–Dietz syndrome (LDS; formerly known as LDS type II) can mimic that of vascular EDS, with overlapping features including translucent skin, easy bruising, atrophic scarring, joint hypermobility, and arterial rupture (see Table 95.5 ). However, some patients with LDS have additional features such as a bifid uvula, hypertelorism, and facial milia. Patients with vascular EDS and other types that result in extensive bruising are often suspected to have a bleeding disorder, and they may present to a dermatologist after a hematologic evaluation fails to reveal any abnormalities.
The brittle cornea syndrome is a rare autosomal recessive condition that has phenotypic overlap with kyphoscoliotic-type EDS, with ocular fragility, blue sclerae, and keratoconus in association with scoliosis, joint hypermobility, and variable skin findings that may include acral wrinkling as well as hyperextensibility and fragility (see Table 95.5 ).
Treatment
The most important intervention is prevention of trauma to the skin, such as the use of shin guards and avoidance of contact sports. Delaying suture removal for up to twice as long as usual plus use of sutures with prolonged tensile strength may improve healing and the appearance of surgical scars.
In vascular EDS, affected individuals are at high risk of arterial and bowel ruptures. Use of celiprolol, a long-acting β 1 -antagonist with partial β 2 -agonist properties, may reduce the incidence of arterial dissection . Approximately 5% of pregnancies in women with vascular EDS are complicated by fatal uterine or arterial rupture ; it is unclear whether elective caesarean section reduces these risks, and patients should be managed in a high-risk obstetric unit with multidisciplinary input.
Pseudoxanthoma Elasticum
▪ Grönblad–Strandberg syndrome
- ■
An autosomal recessive disorder characterized by clumped, distorted, and calcified elastic fibers
- ■
Cutaneous features include yellowish papules, “cobblestoning,” and redundant folds in flexural areas
- ■
Calcification of elastic fibers in the walls of medium-sized arteries can lead to luminal narrowing and clinical sequelae such as claudication, renovascular hypertension, angina, and myocardial infarctions
- ■
Angioid streaks reflect breaks in the calcified elastic lamina of Bruch’s membrane; choroidal neovascularization and hemorrhage can result in loss of visual acuity
- ■
Gastrointestinal hemorrhage is another possible complication
History
The French dermatologist Balzer first described the skin manifestations of PXE and associated elastic degeneration in the skin and heart in 1884 , which was followed 5 years later by a similar case report by Chauffard . Based on these two cases, Darier coined the name “pseudo-xanthome élastique” in 1896 to differentiate this condition from xanthomas . In the late 1920s, Grönblad and Strandberg recognized the association of the characteristic skin manifestations with angioid streaks, and the cardiovascular manifestations were better defined two decades later. More recently, the molecular defects that underlie PXE were identified, clarifying the mode of inheritance, which had previously been debated .
Epidemiology
PXE is an autosomal recessive disease, with a prevalence estimated to be from 1 in 25 000 to 1 in 100 000. There is no racial predilection but a slight female preponderance . Skin changes of PXE usually appear during childhood, but the diagnosis is frequently not made until serious systemic or ocular complications develop in the third or fourth decade of life. Mild cutaneous and ocular findings have also been observed in heterozygous carriers .
Pathogenesis
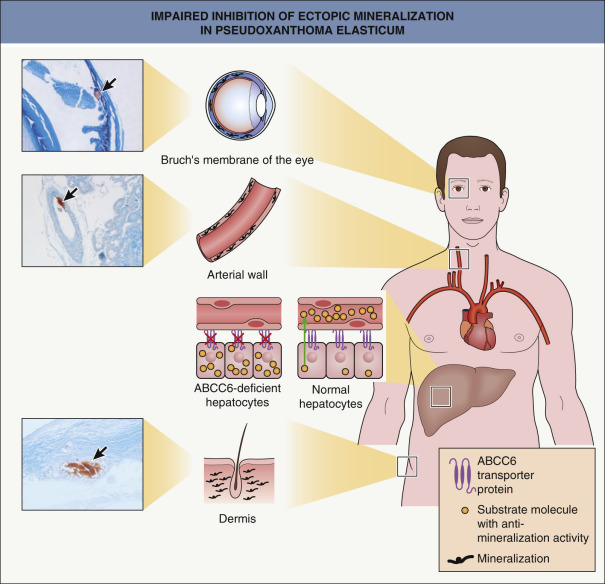
Loss-of-function mutations in both copies of ABCC6 can be identified in most patients with PXE. ABCC6 encodes an ATP-binding cassette (ABC) transporter with a high degree of homology to the multidrug resistance-associated proteins. ABCC6 is predominantly expressed in the basolateral membrane of hepatocytes, where it serves as an efflux pump. The absence of functional ABCC6 in PXE patients results in reduced ATP secretion from hepatocytes, which leads to reduced plasma concentrations of a mineralization inhibitor – inorganic pyrophosphate (PPi) . Serum levels of fetuin A, a major anti-mineralization protein secreted from the liver, are also significantly reduced in PXE patients . Expression of other systemic inhibitors of mineralization such as nucleotidase-5′ ecto (NT5E; converts AMP to adenosine) and osteopontin may also be down-regulated, while expression of mineralization-promoting alkaline phosphatase is upregulated .
Biallelic mutations in ENPP1 , which encodes the ectonucleotide pyrophosphatase/phosphodiesterase 1 enzyme that regulates production of PPi, can cause generalized arterial calcification of infancy (GACI), a rare autosomal recessive disorder that is usually lethal during the first six months of life. However, biallelic ENPP1 mutations occasionally cause PXE, and mutations in ABCC6 underlie a subset of GACI, thus demonstrating pathogenic and phenotypic overlap between the two conditions .
Biallelic mutations in the γ-glutamyl carboxylase gene ( GGCX ) cause PXE-like skin changes plus deficiency in vitamin K-dependent coagulation factors . A combination of heterozygous mutations in GGCX and ABCC6 has also been identified in patients with cutaneous findings of PXE . The GGCX enzyme catalyzes the vitamin K-dependent γ-glutamyl carboxylation that is necessary for activation of both coagulation factors in the liver and the matrix gla protein in peripheral connective tissues. When carboxylated (activated), matrix gla protein prevents ectopic mineralization.
The pathogenesis of PXE and related conditions is therefore related to decreased anti-mineralization activity. Calcification of elastic fibers in the mid and deep dermis, the media and intima of mid-sized arteries, and Bruch’s membrane of the eye produces the characteristic clinical and histopathologic changes of PXE ( Fig. 97.5 ).