Abstract
Allogeneic hematopoietic stem cell transplantation (HSCT) is a potentially curative therapy for a variety of malignant and nonmalignant conditions. However, its use is limited by the risk of graft-versus-host disease (GVHD), the primary cause of morbidity and non-relapse-related mortality in patients who have undergone allogeneic HSCT. Acute GVHD manifests as a cutaneous exanthem, cholestasis, transaminitis, and diarrhea. Chronic GVHD is a multisystem disorder that may affect nearly any organ; the most common sites of involvement are the skin, oral mucosa, and eyes. Chronic cutaneous GVHD is remarkably variable in its clinical presentation, and may resemble lichen planus, lichen sclerosus, morphea, systemic sclerosis, and eosinophilic fasciitis.
Keywords
allogeneic hematopoietic stem cell transplant (HSCT), graft-versus-host disease (GVHD), acute GVHD (aGVHD), chronic GVHD (cGVHD), myeloablative, fasciitis, engraftment syndrome
- ▪
Currently, over 25 000 allogeneic hematopoietic stem cell transplants (HSCTs) are performed worldwide each year
- ▪
Trends in the use of unrelated donors, non-myeloablative conditioning protocols, and donor lymphocyte infusions are impacting the incidence and presentation of graft-versus-host disease (GVHD)
- ▪
Acute GVHD is a major complication of allogeneic HSCT; severe skin involvement, although uncommon, is associated with a high mortality rate
- ▪
Chronic cutaneous GVHD is a polymorphous condition with features resembling lichen planus, lichen sclerosus, morphea and eosinophilic fasciitis
- ▪
Chronic cutaneous GVHD is difficult to treat and the aggressiveness of treatment must be balanced against the complications of chronic immunosuppression
Introduction
Graft-versus-host disease (GVHD) is a multi-organ disorder most commonly due to the transfer of foreign donor hematopoietic stem cells into a host recipient via an allogeneic hematopoietic stem cell transplant (HSCT). GVHD may also occur following transfusion of non-irradiated blood products (to immunocompromised hosts), by maternal–fetal transmission, or in the setting of solid organ transplantation. Despite several decades of experience with HSCT for a broadening array of disorders ( Table 52.1 ), GVHD remains a treatment dilemma and a significant cause of non-relapse-related morbidity and mortality. Acute GVHD (aGVHD) often presents in a dramatic fashion and requires prompt diagnosis and treatment. Management of chronic GVHD (cGVHD), particularly in patients who are refractory to first-line therapy, remains challenging.
| PRIMARY DISORDERS TREATED WITH ALLOGENEIC HEMATOPOIETIC CELL TRANSPLANTATION |
|
History
The first successful allogeneic bone marrow transplant was performed in 1968. Subsequent advances in histocompatibility antigen testing increased safety, while establishment of bone marrow donor registries made HSCT accessible to thousands of patients each year who lacked a suitable related donor. However, use of matched unrelated donor (MUD) transplants carries a greater likelihood of a mismatch of minor human leukocyte antigen (HLA) loci compared to matched related donors, which may be partially responsible for the rising incidence of cGVHD. More recently, the use of umbilical cord blood and reduced-intensity conditioning regimens has led to a further increase in the number of transplants. The former increases the number of potential donors for those who have no related or unrelated matched donors while the latter allows those patients unable to tolerate myeloablative conditioning to receive an HSCT.
Epidemiology
Currently, over 25 000 allogeneic HSCTs are performed worldwide each year. Although a variety of donor and recipient factors ultimately impact GVHD incidence (e.g. older age, T-cell-replete graft), the most important predictor of GVHD remains HLA compatibility between donor and recipient. Approximately 40% of HLA-identical HSCT recipients and 60–70% of HLA-mismatched HSCT recipients will develop GVHD.
Increasingly, umbilical cord blood and peripheral blood are utilized as the stem cell source in place of bone marrow. Cord blood transplantation is associated with a decreased incidence of GVHD, but higher rates of non-engraftment. In addition, because the number of stem cells present in cord blood is limited, two cord blood units are often used for successful engraftment into an adult recipient. Peripheral blood has clearly emerged as the preferred stem cell source at many major transplant centers. In peripheral blood HSCTs, the donor is treated with a colony-stimulating factor (e.g. filgrastim) which mobilizes the donor stem cells from the marrow into the circulation. Cells are then collected via apheresis, processed, and infused into the recipient. Peripheral blood HSCT is associated with more rapid engraftment when compared to bone marrow HSCT; however, there is an increased risk of cGVHD .
Several other trends in HSCT are affecting the natural history of GVHD. Removal of donor T cells (“T-cell depletion”) prior to transplantation through ex vivo graft manipulation (e.g. cell separation), or in vivo treatment of the recipient (e.g. antithymocyte globulin, alemtuzumab), significantly decreases the risk of GVHD, but at the expense of increased cancer relapse due to abrogation of donor T-cell graft-versus-tumor (GVT) effect. Recognition of the key role of GVT effect (rather than the conditioning chemotherapy) in cancer remission has also led to the proliferation of non-myeloablative and reduced-intensity conditioning regimens. Although lower doses of chemotherapy and/or radiation are utilized, they are sufficiently immunodepleting to allow engraftment. This minimizes conditioning-related toxicities (e.g. mucositis) and allows for HSCT in older patients and those with comorbidities. While these regimens appear to decrease the risk of aGVHD, they may also delay the appearance of “classic” aGVHD manifestations. The use of total body irradiation for conditioning (in place of or in conjunction with chemotherapy) may also confer an increased risk for the later development of fibrosing skin manifestations of cGVHD . Finally, donor lymphocyte infusions (DLI), administered to the recipient post-HSCT to augment the GVT effect, have altered the traditional timing of both “acute” and “chronic” disease. For instance, DLI may induce classic aGVHD manifestations even when administered after the 100-day time period.
The skin is the most frequently affected organ in both aGVHD and cGVHD. Approximately 80% of patients who develop aGVHD have skin involvement at the time of diagnosis ; however, prevalence rates vary dramatically between HSCT protocols, and differentiation of sclerotic versus non-sclerotic features are often not reported, rendering estimates of specific manifestations difficult. In the cGVHD setting, non-sclerotic skin disease generally presents earlier than sclerotic disease. However, neither antecedent aGVHD nor “lichenoid” cGVHD skin involvement is a prerequisite to the later development of sclerotic disease. In one institutional review of 270 consecutive patients following HLA-identical HSCT, only 7 (13%) of 53 patients with cGVHD manifested sclerotic features . In contrast, in a cross-sectional cohort of 206 NIH cGVHD patients enriched for refractory disease, sclerotic features were detected in 109 (53%) .
Pathogenesis
The pathogenesis of aGVHD can be summarized in a three-step process. First, HSCT conditioning and consequent damage to host tissues leads to activation of host antigen-presenting cells (APC). Second, donor T cells proliferate in response to contact with activated APC. Finally, destruction of target tissues (skin, liver, gastrointestinal tract) occurs via cytotoxic T lymphocytes, natural killer cells, and soluble factors (TNF, IFN-γ, IL-1, nitric oxide) .
By contrast, a clear understanding of cGVHD is still lacking and available murine models recapitulate only select features of human disease (e.g. sclerotic skin involvement, autoantibodies). Alloreactive T cells are again thought to play a central role, but a number of other immune mediators have been increasingly implicated. Many cGVHD manifestations resemble autoimmune disease, suggesting a role for B cells in the disease process. Indeed, rituximab (anti-CD20 antibody) is a beneficial salvage therapy in some patients. B cells appear to prime T cells to minor histocompatibility antigens, and levels of B -cell a ctivating f actor of the TNF f amily (BAFF), a marker of B-cell activation, correlate with cGVHD. Multiple autoantibodies are often present in patients with cGVHD, including antinuclear, anti-dsDNA, and anti-smooth muscle antibodies, although, to date, they have not been shown to correlate with specific disease manifestations. Activating antibodies against the platelet-derived growth factor receptor (PDGFR) were observed in patients with cGVHD, suggesting that targeting this receptor and its profibrotic pathway with agents such as imatinib mesylate could improve cGVHD . However, the functional significance of these antibodies remains uncertain .
Characterization of immunologic differences between “early” and “late” cGVHD may provide important insight into the phenotypic variety of cGVHD. Early-onset cGVHD (3–9 months) is associated with IFN-γ, increased regulatory T cells, and a T-cell cytokine response (IL-2Rα). By contrast, late cGVHD (>9 months) is characterized by lack of a Th2 shift, B-cell activation via soluble BAFF, induction of Toll-like receptor 9 (TLR9) highly-expressing B cells, and autoantibody formation . In chronic lichenoid GVHD, a mixed Th1/Th17 signature with elevated IFN-γ and IL-17-producing T cells suggests that newly available agents targeting these signaling pathways may provide novel avenues of treatment . Recent data from an IL-17-dependent sclerodermatous cGVHD model also suggest that skin fibrosis may be driven by TGF-β via donor-derived, skin-infiltrating macrophages . Careful definitions of disease states, in particular distinction between sclerotic and non-sclerotic skin manifestations, will be invaluable for validation of new therapies.
Clinical Features
Acute GVHD
Acute GVHD presents in the skin as a morbilliform exanthem ( Fig. 52.1 ), with an initial predilection for acral areas (e.g. dorsal hands and feet, palms, soles, forearms, ears), as well as the upper trunk. Lesions most commonly appear 4–6 weeks following HSCT despite prophylactic immunosuppressive therapy. Pruritus is variable and a folliculocentric pattern may be observed. Patients may be thrombocytopenic, which can give the eruption a hemorrhagic appearance. The gastrointestinal tract (nausea, voluminous diarrhea, abdominal pain) and the liver (transaminitis, cholestasis, bilirubin elevation) are the two other primary organ systems affected by aGVHD. Together with body surface area of skin involvement, these manifestations are used for staging and grading of disease severity ( Table 52.2 ). Stage IV acute skin disease consists of generalized involvement with bullae formation which may lead to skin sloughing resembling toxic epidermal necrolysis and portends a very low likelihood of survival.
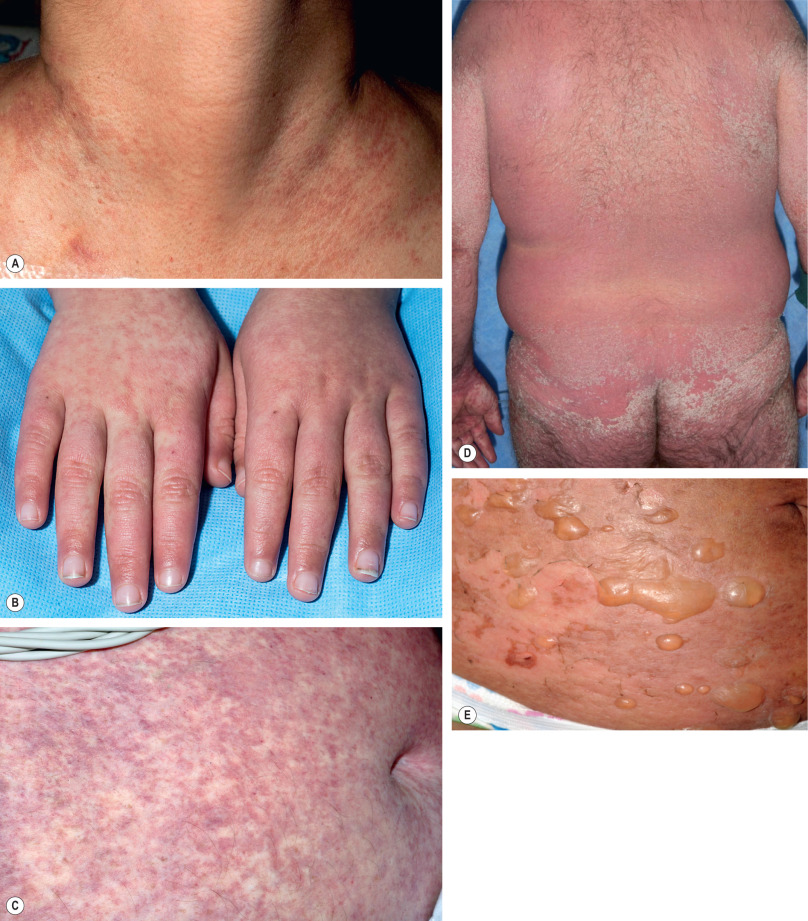
| CLINICAL STAGING AND HISTOLOGIC GRADING OF ACUTE GRAFT-VERSUS-HOST DISEASE | |||||
|---|---|---|---|---|---|
| Stage | Skin | Liver | Gut | Grade | Histologic |
| 1 | Erythematous macules and papules, <25% BSA | Bilirubin 2 to <3 mg/dl | Diarrhea, 500–1000 ml/day, or persistent nausea | I | Focal vacuolar change of basal keratinocytes |
| 2 | Erythematous macules and papules, 25–50% BSA | Bilirubin 3–6 mg/dl | Diarrhea, 1000–1500 ml/day | II | Grade I plus necrotic keratinocytes in the epidermis and/or hair follicle and a dermal lymphocytic infiltrate |
| 3 | Erythematous macules and papules (>50% BSA) to generalized erythroderma | Bilirubin 6–15 mg/dl | Diarrhea, >1500 ml/day | III | Grade II plus fusion of basilar vacuoles to form clefts and microvesicles |
| 4 | Generalized erythroderma with bulla formation | Bilirubin >15 mg/dl | Severe abdominal pain with or without ileus | IV | Grade III plus large areas of separation of epidermis from dermis |
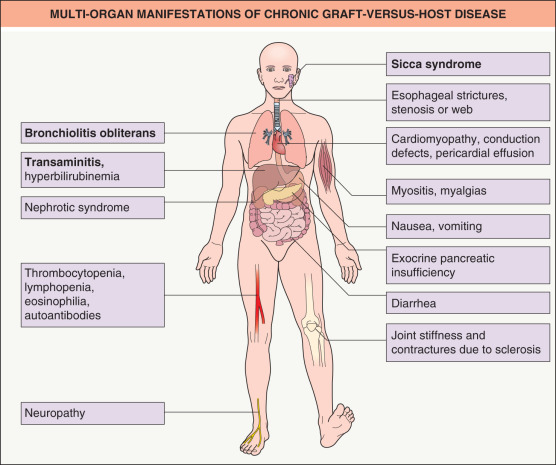
Chronic GVHD
In contrast to aGVHD, cGVHD may manifest in nearly any organ system ( Fig. 52.2 ). Skin and mucosal involvement are exceedingly common, but there is remarkable variability in both presentation and severity. Other common sites of involvement include the eyes (keratoconjunctivitis sicca, blepharitis, corneal erosions), salivary glands (sicca syndrome), and lungs (bronchiolitis obliterans); less commonly, the esophagus (strictures, webs), liver, and pancreas (exocrine insufficiency) are affected.
In the past, differentiation of aGVHD and cGVHD was linked to time of onset following HSCT (≤ or > 100 days); however, this is a somewhat arbitrary distinction and changing HSCT regimens have altered the onset of classic aGVHD and cGVHD manifestations. For instance, features of both aGVHD and cGVHD may occur simultaneously (overlap chronic GVHD), or acute features may first present after day 100 (delayed acute GVHD). The NIH Consensus Project proposed a classification of cGVHD using organ-specific criteria . “Diagnostic” criteria, i.e. those skin manifestations sufficient to make a clinical diagnosis of cGVHD, are provided in Table 52.3 . “Distinctive” cGVHD criteria (e.g. papulosquamous lesions, alopecia) are those which require biopsy confirmation as well as exclusion of other possible etiologies in order for a diagnosis of cGVHD to be established.
| DIAGNOSTIC MUCOCUTANEOUS MANIFESTATIONS OF CHRONIC GRAFT-VERSUS-HOST DISEASE |
| Skin |
|
| Oral mucosa |
|
| Genital mucosa |
|
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


