Abstract
Advances in molecular technology have led to elucidation of the genetic bases of many single-gene inherited and mosaic skin disorders, greatly improving our understanding of these conditions. Genotype–phenotype correlations in genodermatoses are often complex, with multiple examples of allelic and locus heterogeneity. Molecular classification of genodermatoses into categories such as keratin defects and RASopathies complements traditional morphologic classification, highlighting pathomechanisms and relationships between conditions. Determining the molecular basis of monogenic skin disorders can enable the development of targeted therapies and provide insights into the pathogenesis of acquired skin diseases.
Keywords
genodermatoses, genotype, phenotype, molecular classification, contiguous gene syndromes, mosaicism, revertant mosaicism, chromosomal disorders, prenatal diagnosis
- ▪
Advances in molecular technology have led to elucidation of the genetic bases of many single-gene inherited and mosaic skin disorders, greatly improving our understanding of these conditions
- ▪
McKusick’s Online Mendelian Inheritance in Man (OMIM) database provides easily accessible, up-to-date information on human genes and genetic diseases; each gene or phenotype entry is assigned a specific six-digit MIM number
- ▪
Genotype–phenotype correlations in genodermatoses are often complex, with multiple examples of allelic heterogeneity (mutations in a single gene causing more than one disorder) and locus heterogeneity (mutations in different genes causing the same disorder)
- ▪
Molecular classification of genodermatoses into categories such as keratin defects and RASopathies complements traditional morphologic classification, highlighting pathomechanisms and relationships between conditions
- ▪
Certain syndromic associations actually represent contiguous gene syndromes caused by large deletions that affect two or more neighboring genes
- ▪
Types 1 and 2 mosaicism in autosomal dominant disorders have been confirmed on a molecular level, and functional X-chromosome mosaicism can lead to a mosaic distribution of skin lesions in female patients heterozygous for X-linked disorders
- ▪
Determining the molecular basis of monogenic skin disorders can enable the development of targeted therapies and provide insights into the pathogenesis of acquired skin disease
- ▪
Molecular research has begun to pave the way to the ultimate goal of gene therapy for severe inherited skin disorders such as epidermolysis bullosa
Introduction
In 1987, deletions in the steroid sulfatase gene were found to underlie X-linked recessive ichthyosis . This heralded an era of tremendous progress in the elucidation of the genetic bases of inherited skin disorders, made possible by rapid advances in molecular technology (including the development of next-generation massively parallel sequencing), discovery of candidate genes, utilization of animal models, and sequencing of the human genome, including single nucleotide polymorphisms (SNPs) useful for linkage analysis and homozygosity mapping (see Ch. 54 ) . Better understanding of signaling pathways, molecules involved in cell–cell communication and adhesion, and mechanisms of cutaneous differentiation have also contributed to the momentum.
As a result of this explosion in research, more than 1000 genes were recognized to be responsible for a particular human phenotype by the year 2000, with approximately 300 of these conditions including cutaneous abnormalities ; by 2017, these numbers had increased to nearly 3800 and 1400, respectively . Currently, the molecular genetic bases of the majority of single-gene inherited skin disorders have been established . The emergence of new genomic and proteomic databases has transformed laborious positional cloning approaches and traditional functional studies, circumventing previous obstacles to the study of rare conditions and facilitating identification of candidate genes. For example, microarray (see Ch. 3 )-based SNP genotyping enabled the identification of ABCA12 mutations as the cause of harlequin ichthyosis; although classic linkage analysis was not possible due to the limited size of the pedigrees, homozygosity mapping (utilizing the aforementioned technology) linked this autosomal recessive condition to a region of homozygosity shared by affected individuals from diverse ethnic backgrounds . With whole-exome (the transcribed portion of the genome) and whole-genome sequencing now feasible and relatively affordable, disease-causing genes can potentially be identified without genetic mapping .
In the past, diagnosis of genodermatoses was complicated by the existence of multiple complex classification systems based on various combinations of clinical, histologic, radiographic, and biochemical criteria. Inconsistent nomenclature laden with descriptive terms, eponyms, and synonyms added to the confusion and potential for misdiagnosis . As the genetic bases of genodermatoses have been determined, integration of molecular and clinical data has helped to simplify disease categorization and eliminate redundant terminology. This has been successfully accomplished for disorders such as epidermolysis bullosa (EB) and ichthyoses, but it represents a work in progress, to be continually refined as additional genotype–phenotype correlations are established . In addition, grouping hereditary skin disorders according to their molecular bases ( Table 55.1 ) can supplement traditional morphologic classification, clarifying pathomechanisms and the relationships between conditions.
| MOLECULAR CLASSIFICATION OF GENETIC SKIN DISORDERS | |
|---|---|
| Molecular defect | Examples of skin disorders that result |
| Keratin defects | See Table 56.4 and Fig. 56.5 |
| Defects in structural proteins of the cornified cell envelope | Ichthyosis vulgaris, loricrin keratoderma (variant Vohwinkel syndrome) |
| Lipid metabolism defects | Various ichthyoses, including CHILD syndrome, Conradi–Hünermann–Happle syndrome, neutral lipid storage disease, lamellar ichthyosis/congenital ichthyosiform erythroderma spectrum (e.g. defective lipoxygenase 3 or 12R, cytochrome p450 4F22), self-healing collodion baby (defective lipoxygenase 3 or 12R), late-onset autosomal recessive congenital ichthyosis, Refsum disease, rhizomelic chondrodysplasia punctata, Sjögren–Larsson syndrome, X-linked recessive ichthyosis and ichthyosis, intellectual disability and spastic quadriplegia; various hyperlipidemias; Farber lipogranulomatosis, Gaucher disease type 2, hyperimmunoglobulinemia D syndrome, localized autosomal recessive hypotrichosis II/autosomal recessive woolly hair, Niemann–Pick disease, psoriasiform dermatitis–microcephaly–developmental delay syndrome |
| Transglutaminase defects | Lamellar ichthyosis/congenital ichthyosiform erythroderma spectrum (defective transglutaminase 1), acral peeling skin syndrome |
| Protease/proteasome defects | Dermatosparaxis, familial hidradenitis suppurativa, hereditary angioedema (type III), Howel-Evans syndrome, ichthyosis–hypotrichosis syndrome, IFAP ( i chthyosis f ollicularis– a trichia– p hotophobia) syndrome, KLICK ( k eratosis l inearis– i chthyosis c ongenita–sclerosing k eratoderma) syndrome, Olmsted syndrome, Papillon–Lefèvre and Haim–Munk syndromes, prolidase deficiency, restrictive dermopathy (defective zinc metallopeptidase), hereditary thrombotic thrombocytopenic purpura, MONA ( m ulticentric o steolysis, n odulosis, and a rthropathy), proteasome-associated autoinflammatory/CANDLE/Nakajo-Nishimura syndrome) |
| Protease inhibitor defects | Antithrombin III deficiency, autosomal recessive exfoliative ichthyosis, hereditary angioedema (types I & II), Netherton syndrome |
| Desmosomal defects (see Table 56.5 and Fig. 56.8 ) | Acantholytic epidermolysis bullosa simplex, arrhythmogenic right ventricular dysplasia/cardiomyopathy + palmoplantar keratoderma + woolly hair; Carvajal syndrome, erythrokeratodermia-cardiomyopathy syndrome, generalized inflammatory peeling skin syndrome, hypotrichosis and skin lesions, hypotrichosis simplex of the scalp, localized autosomal recessive hypotrichosis, monilethrix (autosomal recessive), Naxos disease, SAM ( s evere dermatitis, a llergies, and m etabolic wasting), skin fragility-ectodermal dysplasia syndrome, skin fragility/woolly hair syndrome, striate palmoplantar keratoderma |
| Connexin defects (see Table 58.5 ) | Bart–Pumphrey syndrome, Clouston syndrome, erythrokeratodermia variabilis, hereditary lymphedema type IC, ILVEN, keratoderma-hypotrichosis-leukonychia totalis, KID ( k eratitis– i chthyosis– d eafness) syndrome, oculodentodigital dysplasia, palmoplantar keratoderma with deafness, porokeratotic adnexal ostial nevus, Vohwinkel syndrome (classic) |
| Other defects in cell-to-cell adhesion | Ichthyosis–hypotrichosis–sclerosing cholangitis syndrome, cleft lip/palate–ectodermal dysplasia syndrome, ectodermal dysplasia–syndactyly syndrome, ectodermal dysplasia–ectrodactyly–macular dystrophy, hypotrichosis with juvenile macular dystrophy, leukocyte adhesion deficiency type I |
| Defects in keratinocyte–extracellular matrix (ECM) adhesion | Various forms of epidermolysis bullosa (EB) simplex, junctional EB, and dystrophic EB (keratin–ECM linkage); Kindler syndrome (actin–ECM linkage) |
| ATP-binding cassette (ABC) transporter defects | Harlequin ichthyosis, lamellar ichthyosis/congenital ichthyosiform erythroderma spectrum (defective ABCA12 ), pseudoxanthoma elasticum, sitosterolemia, Tangier disease/familial hypoalphaproteinemia |
| Calcium pump defects | Darier disease, Hailey–Hailey disease |
| Copper transporter defects | Menkes disease, occipital horn syndrome, Wilson disease |
| Defects in other transporters | Acrodermatitis enteropathica (zinc transporter), arterial tortuosity syndrome (glucose transporter), autosomal dominant hemochromatosis (iron-regulated transporter), spondylodysplastic Ehlers–Danlos syndrome (zinc transporter), H syndrome (nucleoside transporter), Hartnup disease (neutral amino acid transporter), ichthyosis–prematurity syndrome (fatty acid transporter), oculocutaneous albinism (transporters on lysosome-related organelles) |
| Collagen defects | Various types of dystrophic EB, various types of Ehlers–Danlos syndrome, Ullrich muscular dystrophy, lysyl hydroxylase 3 deficiency |
| Defects in other ECM proteins | Autosomal dominant cutis laxa, autosomal recessive cutis laxa type I, cutis laxa with severe pulmonary/gastrointestinal/urinary abnormalities, classic-like and hypermobile Ehlers–Danlos syndrome, juvenile hyaline fibromatosis/infantile systemic hyalinosis, lipoid proteinosis, Marfan syndrome, stiff skin syndrome |
| Nuclear membrane defects (see Table 63.10 ) | Buschke–Ollendorff syndrome/melorheostosis, Néstor–Guillermo progeria syndrome, Hutchinson–Gilford progeria syndrome, mandibuloacral dysplasia, partial > generalized lipodystrophy (familial and “acquired” forms), restrictive dermopathy |
| Defects in pyrin/NOD-family members and related proteins (see Fig. 4.2 ) | Blau syndrome, NOMID syndrome, familial cold autoinflammatory syndrome, familial Mediterranean fever, Muckle–Wells syndrome, PAPA syndrome |
| Interferonopathies (see Ch. 45 ) | Aicardi-Goutières syndrome, familial chilblain lupus, proteasome-associated autoinflammatory/CANDLE/Nakajo-Nishimura syndrome, SAVI ( S TING- a ssociated v asculopathy with onset in i nfancy), X-linked reticulate pigmentary disorder |
| RecQ DNA helicase defects | Bloom syndrome, Rothmund–Thomson syndrome, Werner syndrome |
| Classic DNA repair defects | Various subtypes of xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome; Muir–Torre syndrome, constitutional mismatch repair deficiency syndrome |
| Cell cycle checkpoint defects | Ataxia telangiectasia, familial atypical mole and melanoma (FAMM) syndrome, multiple endocrine neoplasia (MEN) 4 |
| Defects in telomere maintenance | Dyskeratosis congenita (see Table 67.8 ; 11 genes implicated to date) |
| RAS–MAPK pathway activation (RASopathies; see Fig. 55.4 ) | Capillary malformation–arteriovenous malformation, cerebral capillary malformations (familial), cardio-facio-cutaneous syndrome, congenital melanocytic nevi, Costello syndrome, Legius syndrome, neurofibromatosis type 1, Noonan syndrome (with multiple lentigines), Noonan-like disorder with loose anagen hair, verrucous epidermal nevi, nevus sebaceus, woolly hair nevi |
| Phosphatidylinositol 3-kinase (PI3K)/AKT pathway activation | Epidermal nevi, lymphatic malformations, PIK3CA-related overgrowth spectrum (see Table 104.5 ), Proteus syndrome, PTEN-hamartoma tumor syndrome, tuberous sclerosis complex, venous malformations |
| G protein activation | Diffuse capillary malformation with overgrowth (some patients), extensive dermal melanocytosis, McCune-Albright syndrome (versus loss-of-function in Albright hereditary osteodystrophy), phakomatosis pigmentovascularis, port-wine stains, Sturge-Weber syndrome |
| cAMP and AMP-activated protein kinase pathway defects (see Fig. 55.3 ) | Albright hereditary osteodystrophy, Carney complex, McCune–Albright syndrome, Peutz–Jeghers syndrome, tuberous sclerosis complex |
| Defects in WNT/β-catenin signaling (see Fig. 55.6 ) | Anonychia congenita, ectodermal dysplasia–neoplastic syndrome, Gardner syndrome, Goltz syndrome (focal dermal hypoplasia), hereditary hypotrichosis simplex, palmoplantar keratoderma with cutaneous SCC and sex reversal, WNT10A-related ectodermal dysplasias (including odonto-onycho-dermal dysplasia and Schöpf–Schulz–Passarge syndrome) |
| Defects in transforming growth factor (TGF)-β signaling | Buschke–Ollendorff syndrome/melorheostosis, Ferguson–Smith multiple self-healing squamous epitheliomas, hereditary hemorrhagic telangiectasia, Loeys–Dietz syndrome, Marfan syndrome, stiff skin syndrome |
| Defective vesicle assembly/protein sorting to vesicles | ARC ( a rthrogryposis– r enal d ysfunction– c holestasis) syndrome, Hermansky–Pudlak syndrome, MEDNIK ( m ental retardation, e nteropathy, d eafness, n europathy, i chthyosis, k eratodermia) syndrome |
| Defective vesicle trafficking or transport | Autosomal recessive cutis laxa type IIA, CEDNIK ( ce rebral d ysgenesis, n europathy, i chthyosis and k eratoderma) syndrome, Chédiak–Higashi syndrome, gerodermia osteodysplastica, Griscelli syndrome, ocular albinism type 1, wrinkly skin syndrome, MACS ( m acrocephaly, a lopecia, c utis laxa and s coliosis) syndrome |
As the “morbid anatomy of the dermatologic genome” continues to be established, new challenges will arise and additional questions will be answered . Because of the rarity of many genodermatoses, their full clinical spectra have yet to be elucidated. However, with the increasing availability of genetic and biochemical testing ( www.genetests.org ), dermatologists can establish the diagnosis in patients with mild or atypical presentations, thereby expanding the range of phenotypes. Continuing to decipher heritable skin disorders will require close interactions between basic scientists and clinicians . Hopefully, such translational research will continue to lead to better understanding of cutaneous structure and function; insights into the pathogenesis of common multifactorial disorders; and effective therapies as well as diagnostic and prognostic information, improved genetic counseling and DNA-based prenatal/preimplantation testing for patients with genodermatoses.
In this chapter, Tables 55.3 to 55.8 divide selected monogenic skin disorders with a known genetic basis into categories according to their primary clinical features. Groups of monogenic conditions similarly listed in tables elsewhere in the book are noted in Table 55.2 .
| GROUPS OF MONOGENIC SKIN DISORDERS LISTED IN TABLES ELSEWHERE IN THE BOOK | |
| Category | Table(s) |
| Congenital insensitivity to pain and related neuropathies | 6.10 |
| Epidermolysis bullosa and other blistering disorders | 32.1 & 32.3 |
| Sclerodermoid disorders | 43.7 |
| Autoinflammatory diseases | 45.2 & 45.7 |
| Porphyrias | 49.3 |
| Keratinopathies and desmosomal disorders | 56.4 ; Figs 56.5 & 56.8 |
| Ichthyoses | 57.1 & 57.4 |
| Connexin disorders and keratodermas | 58.5 & 58.6 |
| Primary immunodeficiencies | 60.4 , 60.6 , 60.8 , 60.10 , 60.13 , 60.15 , & 60.16 |
| Disorders associated with multiple café-au-lait macules | 61.4 |
| Epidermal nevi and other mosaic disorders | 62.4 & 62.7 |
| Progeroid syndromes/inherited poikilodermas and nuclear membrane disorders | 63.9 & 63.10 |
| Ectodermal dysplasias | 63.11 , 63.12 , 63.13 |
| Aplasia cutis congenita, cleft lip/palate, and digital anomalies | 64.3 , 63.4 , 63.5 , 63.6 |
| Disorders of hypopigmentation (diffuse or circumscribed) | 65.1 , 66.4 |
| Disorders with reticulated hyperpigmentation, dyskeratosis congenita, and dyschromatoses | 67.7 , 67.8 , & 67.10 |
| Hypotrichosis, hair shaft disorders, and hypertrichosis | 69.8 ; 70.1 & 70.2 |
| Xeroderma pigmentosa and other photosensitivity disorders | 86.2 , 87.4 |
| Hyperlipidemias | 92.2 |
| Ehlers–Danlos syndrome, cutis laxa, and other extracellular matrix disorders | 95.5 ; 97.1 , 97.2 , & 97.6 |
| Lipodystrophy syndromes | 101.1 |
| Vascular anomalies and overgrowth syndromes | 104.2 & 104.5 |
| Disorders with multiple lentigines | 112.2 |
| SELECTED HEREDITARY DISORDERS OF THE NAILS | |||
| Disorder | Mode of inheritance | Gene or gene product | Gene symbol |
| Anonychia congenita | AR | R-spondin family, member 4 | RSPO4 |
| Isolated AR nail dysplasia | AR | Frizzled 6 | FZD6 |
| Isolated congenital nail clubbing | AR | Hydroxyprostaglandin dehydrogenase 15-(NAD) | HPGD |
| Isolated toenail dystrophy | AD | Collagen VII, α1 chain | COL7A1 |
| Leukonychia totalis | AD, AR | Phospholipase C delta 1 | PLCD1 |
| Nail–patella syndrome | AD | LIM homeobox transcription factor 1β | LMX1B |
| Pachyonychia congenita-6a | AD | Keratin 6a | KRT6A |
| Pachyonychia congenita-16 | AD | Keratin 16 | KRT 16 |
| Pachyonychia congenita-6b | AD | Keratin 6b | KRT6B |
| Pachonychia congenita-17 | AD | Keratin 17 | KRT17 |
| Tricho-rhino-phalangeal syndrome 1 | AD | TRPS1 (zinc finger protein) | TRPS1 |
| SELECTED HEREDITARY METABOLIC SKIN DISORDERS | ||||
| Disorder | Mode of inheritance | Gene or gene product | Gene symbol | |
| Disorders due to enzyme deficiencies (see Ch. 63 ) | ||||
| Alkaptonuria | AR | Homogentisate 1,2-dioxygenase (homogentisate oxidase) | HGD | |
| Biotinidase deficiency | AR | Biotinidase | BTD | |
| Holocarboxylase synthetase deficiency | AR | Holocarboxylase synthetase | HLCS | |
| Fabry disease * | XR | α-Galactosidase A | GLA | |
| Fucosidosis * | AR | α- l -Fucosidase | FUCA1 | |
| Farber lipogranulomatosis | AR | Acid ceramidase | ASAH | |
| Gaucher disease types I–III | AR | Acid β-glucosidase (glucocerebrosidase) | GBA | |
| Hereditary angioedema | Types I & II | AD | Serpin peptidase inhibitor, clade G, member 1 (C1 esterase inhibitor) | SERPING1 |
| Type III | AD (only females affected) | Factor XII (Hageman factor) | F12 | |
| Homocystinuria | AR | Cystathionine β-synthase † | CBS | |
| Niemann–Pick disease type A | AR | Acid sphingomyelinase | SMPD1 | |
| Phenylketonuria | AR | Phenylalanine hydroxylase | PAH | |
| AR | Quinoid dihydropteridine reductase | QDPR | ||
| AR | 6-Pyruvoyltetrahydropterin synthase | PTS | ||
| Prolidase deficiency | AR | Prolidase (peptidase D) | PEPD | |
| Disorders due to defective transporters/related proteins | ||||
| Acrodermatitis enteropathica | AR | Solute carrier family 39, member 4 (zinc transporter) | SLC39A4 | |
| H syndrome ‡ | AR | Solute carrier family 29, member 3 (nucleoside transporter) | SLC29A3 | |
| Hartnup disease | AR | Solute carrier family 6, member 19 (neutral amino acid transporter) | SLC6A19 | |
| Hemochromatosis | AR | Hemochromatosis | HFE | |
| AR | Transferrin receptor 2 | TFR2 | ||
| AD | Solute carrier family 40, member 1 (ferroportin) | SLC40A1 | ||
| Hemochromatosis, juvenile | AR | Hepcidin antimicrobial peptide | HAMP | |
| AR | Hemojuvelin | HFE2 | ||
| Wilson disease | AR | Cu 2+ -transporting P-type ATPase 7B | ATP7B | |
* Additional metabolic disorders that can present with angiokeratoma corporis diffusum are presented in Table 63.7 .
† Defects in this gene represent the most common cause of homocystinuria.
‡ Including cases reported as pigmented hypertrichotic insulin-dependent diabetes mellitus syndrome and familial histiocytosis syndrome.
| SELECTED HEREDITARY DISORDERS CHARACTERIZED BY BENIGN SKIN TUMORS | |||
| Disorder | Mode of inheritance | Gene or gene product | Gene symbol |
| Familial cylindromatosis; Brooke–Spiegler syndrome; multiple familial trichoepitheliomas | AD | CYLD (deubiquitinating enzyme) | CYLD |
| Familial mastocytosis ± gastrointestinal stromal tumors * | AD | KIT proto-oncogene (stem cell factor receptor) | KIT |
| Leiomyomatosis, cutaneous and uterine (Reed syndrome) † | AD | Fumarate hydratase | FH |
| Lipomatosis, familial multiple | AD | ||
| Lipomatosis, benign symmetric (Madelung disease) ‡ | mt | Mitochondrial tRNA-lysine | MT-TK |
| AR | Mitofusin 2 | MFN2 | |
| AR | Lipase E, hormone sensitive | LIPE | |
| Ferguson–Smith multiple self-healing squamous epitheliomas | AD | Transforming growth factor-β receptor 1 (loss-of-function mutations) | TGFBR1 |
| Neurofibromatosis type 1 | AD | Neurofibromin 1 (GTPase activating protein) | NF1 |
| Neurofibromatosis type 2 | AD | Neurofibromin 2 (merlin) | NF2 |
| Tuberous sclerosis complex | AD | Hamartin | TSC1 |
| AD | Tuberin | TSC2 | |
† Also associated with renal cell carcinoma.
‡ Associated with myoclonic epilepsy with ragged red fibers ( MT-TK mutations), neuropathy ( MFN2 mutations), or partial lipodystrophy and myopathy ( LIPE mutations).
| SELECTED HEREDITARY SKIN DISORDERS ASSOCIATED WITH EXTRACUTANEOUS CANCER | ||||
| Disorder | Mode of inheritance | Gene or gene product | Gene symbol | |
| Ataxia–telangiectasia | AR | Ataxia–telangiectasia mutated (phosphatidylinositol 3-kinase-like serine/threonine protein kinase) | ATM | |
| Birt–Hogg–Dubé syndrome | AD | Folliculin | FLCN | |
| Bloom syndrome | AR | RecQ protein-like 3 (DNA helicase) | BLM (RECQL3) | |
| Costello syndrome (see text) | AD | v-Ha-ras Harvey rat sarcoma viral oncogene homolog | HRAS | |
| AD | v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog | KRAS | ||
| Cowden-like syndrome | AD | Succinate dehydrogenase complex, subunit B, iron sulfur | SDHB | |
| AD | Succinate dehydrogenase complex, subunit D, integral membrane protein | SDHD | ||
| Dyskeratosis congenita (see Table 67.8 ) | ||||
| Ectodermal dysplasia–neoplastic syndrome | AD | Axis inhibitor 2 | AXIN2 | |
| Gardner syndrome | AD | Adenomatous polyposis coli | APC | |
| Howel–Evans syndrome (tylosis–esophageal carcinoma) | AD | Rhomboid 5 homologue 2 | RHBDF2 | |
| Muir–Torre syndrome | AD | MutS homolog 2 (mismatch repair enzyme) | MSH2 | |
| AD | MutS homolog 6 (mismatch repair enzyme) | MSH6 | ||
| AD | MutL homolog 1 (mismatch repair enzyme) | MLH1 | ||
| MUTYH-associated polyposis | AR | MutY homolog | MUTYH | |
| Constitutional mismatch repair deficiency syndrome (childhood tumor syndrome with neurofibromatosis type 1 phenotype) | AR | MutS homolog 2 (mismatch repair enzyme) | MSH2 | |
| AR | MutS homolog 6 (mismatch repair enzyme) | MSH6 | ||
| AR | MutL homolog 1 (mismatch repair enzyme) | MLH1 | ||
| AR | Postmeiotic segregation increased 2 (mismatch repair enzyme) | PMS2 | ||
| Multiple endocrine neoplasia (MEN) | MEN 1 | AD | Menin | MEN1 |
| MEN 2A | AD | Ret proto-oncogene (cysteine-rich extracellular domain) | RET | |
| MEN 2B | AD | Ret proto-oncogene (particularly Met918Thr in substrate recognition pocket) | RET | |
| MEN 4 | AD | Cyclin-dependent kinase inhibitor 1B (p27, Kip1) | CDKN1B | |
| PTEN hamartoma tumor syndrome (including Cowden and Bannayan–Riley–Ruvalcaba syndromes) | AD | Phosphatase and tensin homolog | PTEN | |
| Rothmund–Thomson syndrome | AR | RecQ protein-like 4 (DNA helicase) | RECQ4 | |
| OTHER SELECTED INHERITED SKIN DISORDERS | ||||
| Disorder | Mode of inheritance | Gene or gene product | Gene symbol | |
| Disorders characterized by excessive extracellular matrix protein deposition (also see Table 43.7 ) | ||||
| Inherited systemic hyalinosis (juvenile hyaline fibromatosis, infantile systemic hyalinosis) | AR | Anthrax toxin receptor 2 (capillary morphogenesis protein 2) | ANTXR2 | |
| Lipoid proteinosis | AR | Extracellular matrix protein 1 | ECM1 | |
| Psoriasiform conditions (also see Table 45.7 for pustular psoriasis variants) | ||||
| Psoriasiform dermatitis–microcephaly–developmental delay | AR | Sterol-C4-methyl oxidase-like | SC4MOL | |
| Seborrhea-like dermatitis with psoriasiform elements | AD | Zinc finger protein 750 | ZNF750 | |
| Familial pityriasis rubra pilaris or (pustular) psoriasis | AD | Caspase recruitment domain family member 14 | CARD14 | |
| Craniosynostosis/skeletal dysplasia syndromes | ||||
| Apert syndrome | AD | Fibroblast growth factor receptor 2 | FGFR2 | |
| Beare–Stevenson cutis gyrata syndrome | AD | Fibroblast growth factor receptor 2 | FGFR2 | |
| Crouzon syndrome with acanthosis nigricans; SADDAN; thanatophoric dysplasia | AD | Fibroblast growth factor receptor 3 * | FGFR3 | |
| Oral–facial–digital syndrome | XD | Oral–facial–digital syndrome 1 | OFD1 | |
| Disorders with prominent neurologic abnormalities (also see Table 6.10 ) | ||||
| Cockayne syndrome † | CSA | AR | ERCC8 (transcription-coupled repair protein) | ERCC8 |
| CSB | AR | ERCC6 (transcription-coupled repair protein) | ERCC6 | |
| Cold-induced sweating syndrome | AR | Cytokine receptor-like factor 1 | CRLF1 | |
| AR | Cardiotrophin-like cytokine factor 1 | CLCF1 | ||
| Primary erythromelalgia | AD | Sodium channel, voltage-gated, type IX, α-subunit | SCN9A | |
| Familial primary localized cutaneous amyloidosis | AD | Oncostatin M receptor | OSMR | |
| Interleukin-31 receptor A | IL31RA | |||
| Defects in coagulation or platelet plugging (see Ch. 23 , Table 105.9 ) | ||||
| Activated protein C resistance (factor V Leiden) | AD | Factor V | F5 | |
| Protein C deficiency | AD | Protein C | PROC | |
| Protein S deficiency | AD | Protein S | PROS1 | |
| Antithrombin III deficiency | AD | Serpin peptidase inhibitor C1 (antithrombin III) | SERPINO | |
| Hyperprothrombinemia | AD | Prothrombin, G20210A polymorphism | F2 | |
| ADAMTS13 deficiency-mediated thrombotic microangiopathy (thrombotic thrombocytopenic purpura) | AR | ADAM metallopeptidase with thrombospondin type 1 motif, 13 (von Willebrand factor-cleaving protease) | ADAMTS13 | |
| Disorders with cutaneous calcification or ossification (see Ch. 50 ) | ||||
| Albright hereditary osteodystrophy | AD | Stimulatory G protein, α-subunit (G s α; inactivating mutations) | GNAS | |
| Progressive osseous heteroplasia | AD | Stimulatory G protein, α-subunit (G s α; inactivating mutations) | GNAS | |
| Fibrodysplasia ossificans progressiva | AD | Activin A receptor, type 1 | ACVR1 | |
| Hyperphosphatemic familial tumoral calcinosis | AR | UDP-N-acetyl-αD-galactosamine:polypeptide N-acetylgalactosaminyltransferase 3 | GALNT3 | |
| AR | Fibroblast growth factor 23 | FGF23 | ||
| AR | Klotho | KL | ||
| Normophosphatemic familial tumoral calcinosis | AR | Sterile alpha motif domain-containing 9 | SAMD9 | |
| Pachydermoperiostosis | AR | 15-hydroxyprostaglandin dehydrogenase | HPGD | |
| AR | Solute carrier organic anion transporter, member 2A1 | SLCO2A1 | ||
* Somatic FGFR3 mutations have been found to underlie a subset of epidermal nevi.
† Patients with mutations in ERCC3 (XPB), ERCC2 (XPD) and ERCC5 (XPG) may have phenotypes with features of both xeroderma pigmentosum and Cockayne syndrome (XP/CS).
Mckusick’s Mendelian Inheritance in MAN
McKusick’s Mendelian Inheritance in Man (MIM) database was first published in 1966 as “Catalogs of Autosomal Dominant, Autosomal Recessive and X-linked Phenotypes”. In the 1994 edition, the subtitle was changed to “A Catalog of Human Genes and Genetic Disorders”, reflecting the progress that had been made in the field. Online MIM (OMIM; https://omim.org/ ) has been widely available on the Internet for over 30 years, providing immediate access to current information on human genes and genetic diseases . This database is updated continuously and can be searched by entering a constellation of clinical features as well as the name of a gene or syndrome.
A particular six-digit number (MIM number) is assigned to each OMIM entry. The first digit of the MIM number indicates the mode of inheritance of the corresponding genetic defect: 1 for autosomal dominant (entries before May 1994); 2 for autosomal recessive (entries before May 1994); 3 for X-linked; 4 for Y-linked; 5 for mitochondrial; and 6 for autosomal dominant or recessive (entries after May 1994). Phenotype entries describe the clinical and biochemical features, inheritance, mapping, and molecular genetics of a given disease or trait; a “#” is used to designate those for which the molecular basis is known. Gene entries are marked by a “*”, while gene plus phenotype entries are indicated with a “+”; both are appended with important disease-causing allelic variants (each of which is given a four-digit extension, beginning with .0001).
Genotype–Phenotype Correlations
Genotype–phenotype correlations in genodermatoses are often complex. Mutations in a single gene can cause more than one clinical disorder, a phenomenon referred to as allelic or clinical heterogeneity (see Table 54.2 ). This can result either from the same mutation occurring in patients with different genetic backgrounds or ages at presentation (e.g. identical PTEN mutations leading to Cowden syndrome and Bannayan–Riley–Ruvalcaba syndrome) or from different mutations (e.g. distinct mutations in the gene encoding lamin A/C leading to Hutchinson–Gilford progeria and familial partial lipodystrophy). Mutations that affect different domains of a protein can produce divergent phenotypes; for example, mutations in the sterile alpha motif (SAM) domain of the p63 protein typically cause AEC ( a nkyloblepharon, e ctodermal dysplasia and c left lip/palate) syndrome, while those in the DNA-binding domain give rise to EEC ( e ctrodactyly, e ctodermal dysplasia and c left lip/palate) syndrome. Various mutations in a particular gene can even lead to disorders with different modes of inheritance, such as X-linked dominant incontinentia pigmenti (a male-lethal condition due to a genomic rearrangement resulting in a partial deletion of the n uclear factor-κB e ssential mo dulator [NEMO] gene) and X-linked recessive hypohidrotic ectodermal dysplasia with immunodeficiency (due to milder, “hypomorphic” mutations in the NEMO gene). Conditions found to represent unexpected examples of allelic heterogeneity in the transforming growth factor-β (TGF-β) signaling pathway include mutations in the TGF-β receptor 1 gene ( TGFBR1 ) in Ferguson–Smith multiple self-healing squamous epithelioma (loss-of-function) as well as Loeys–Dietz syndrome (gain-of-function), and fibrillin 1 gene ( FBN1 ) mutations in generalized stiff skin syndrome as well as Marfan syndrome.
The type of mutation can also affect the severity of a genodermatosis. The potentially lethal generalized severe form of junctional EB (JEB, an autosomal recessive disorder) is typically caused by mutations in the LAMB3 gene that lead to a premature termination codon (resulting in a complete absence of the LAMB3 protein), whereas milder forms of JEB are produced by missense or splice-site mutations in the same gene (resulting in a LAMB3 protein with decreased function). In contrast, in autosomal dominant conditions where there is dimerization of the mutated protein product (e.g. the KIT tyrosine kinase receptor that is defective in piebaldism), dominant negative missense mutations often result in more severe disease (via abnormal proteins binding to and disrupting normal proteins) than do mutations that lead to premature termination codons and thereby result in haploinsufficiency (complete loss of half of the proteins).
In other instances, mutations in different genes produce the same clinical disorder; this is referred to as locus or genetic heterogeneity . Locus heterogeneity can occur when mutated proteins serve a similar function (e.g. components of various b iogenesis of l ysosome-related o rganelle c omplexes [BLOCs] in Hermansky–Pudlak syndrome) or interact with one another in a complex (e.g. hamartin and tuberin in tuberous sclerosis), as a ligand and receptor (e.g. endothelin-3 and the endothelin-B receptor in type 4 Waardenburg syndrome), or in a signaling pathway (e.g. various RAS–mitogen-activated protein kinase [MAPK] pathway proteins in cardio-facio-cutaneous syndrome; see below). However, phenotypic differences can arise when these proteins also have distinct functions and/or tissue distributions; for example, the neurologic abnormalities in Griscelli syndrome 1 result from myosin Va expression in neurons, and the immunodeficiency and hemophagocytic syndrome that characterize Griscelli syndrome 2 reflect RAB27A expression in hematopoietic cells.
Molecular Classification of Hereditary Skin Disorders
Molecular classification (see Table 55.1 ) represents a useful approach to the categorization of genodermatoses. Based on the pathogenetic defect rather than the clinical presentation, it can highlight similarities between conditions with seemingly disparate phenotypes (e.g. classic ichthyoses and metabolic disorders such as Gaucher disease, both caused by defects in lipid metabolism) and facilitate better understanding of important cellular pathways (e.g. RAS signaling) and responses to external stimuli (e.g. pyrin/NOD family members that function in innate immunity). Several groups of genodermatoses for which molecular classification has been instructive are discussed below.
Keratin Defects
In 1991, EB simplex (EBS) became the first human disease shown to be caused by intermediate filament mutations. Since that time, molecular defects in keratins have been identified in a diverse group of hereditary disorders affecting the skin, leading to mechanical fragility and/or abnormal keratinization, depending on the layer of the epidermis in which the defective protein is expressed; the hair, nails, and oral mucosa may also be involved (see Table 55.2 ) . Dowling–Degos disease and Naegeli–Franceschetti–Jadassohn (NFJ) syndrome, which are characterized by reticulated pigmentation and (for NFJ) ectodermal dysplasia, represent entities more recently recognized as belonging to the keratin disorder category.
Keratin intermediate filaments, which are expressed in a tissue- and differentiation-specific fashion, are composed of heterodimeric subunits formed by the specific pairing of a type I (acidic; KRT9–20, 25–28 [inner root sheath], 31–40 [hair]) and type II (basic–neutral; KRT1–8, 71–75 [inner root sheath], 81–86 [hair]) keratin (see Ch. 56 ). Keratin genes are clustered at two loci in the human genome, 17q21 (type I keratins) and 12q13 (type II keratins). Many keratin disorders can be caused by mutations (often dominant negative) in either the type I or type II component of a particular keratin pair, thus exhibiting locus heterogeneity (e.g. KRT10 or KRT1 for epidermolytic ichthyosis; KRT14 or KRT5 for EBS). Allelic heterogeneity is also observed, with mutations in the highly conserved helix initiation and termination motifs leading to more severe phenotypes than those in other regions (e.g. the generalized severe form of EBS versus localized EBS) (see Fig. 56.5 ).
Defects in Intercellular Junctions
Mutations in genes encoding components of desmosomes and gap junctions underlie a variety of genodermatoses, several of which have prominent extracutaneous manifestations.
Desmosomal defects
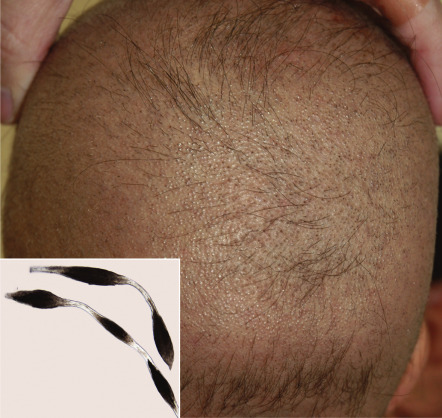
Desmosomes are intercellular junctions that provide mechanical integrity to tissues by anchoring intermediate filaments to the cell surface and mediating strong cell–cell adhesion (see Ch. 56 ). They are particularly important in stratified squamous epithelia and the myocardium, tissues that are subjected to substantial mechanical stress . It is therefore not surprising that impaired keratinization (especially palmoplantar keratoderma), skin fragility, and cardiomyopathy represent features of disorders caused by defects in desmosomal proteins such as desmoplakin, plakoglobin and plakophilin 1 (see Fig. 56.8 and Table 56.5 ). Abnormal hair (oftentimes woolly in nature) can also result from abnormalities in these proteins, and defects in other desmosomal components such as desmoglein 4 and corneodesmosin (which are highly expressed within the hair follicle) can lead to hypotrichosis and (for desmoglein 4) an autosomal recessive form of monilethrix ( Fig. 55.1 ). Additional consequences of desmosomal defects include inflammatory peeling skin (corneodesmosin) and s evere dermatitis, a llergies, and m etabolic wasting (SAM; desmoglein 1, desmoplakin).
Connexin defects
Gap junctions are intercellular channels that connect the cytoplasm of neighboring cells, facilitating communication that coordinates cellular growth, differentiation and responses to stimuli as well as tissue morphogenesis and homeostasis (see Ch. 58 ). Transmembrane connexin proteins undergo oligomerization to form the connexons that compose gap junctions. Connexin proteins such as Cx26, Cx30, Cx31, and Cx43 are preferentially expressed in ectoderm-derived epithelia of the inner ear and cornea as well as in the epidermis and its appendages . This accounts for the sensorineural deafness, keratitis, and cutaneous abnormalities ranging from keratoderma to erythrokeratoderma to ectodermal dysplasias affecting the hair and nails that are observed in various connexin disorders (see Table 55.1 ). Several connexins are expressed in lymphatic cells, and mutations in the gene encoding Cx47 cause a hereditary type of lymphedema .
Defects in Keratinocyte–Extracellular Matrix Adhesion
The hemidesmosome links keratin intermediate filaments (KRT5, KRT14) within basal keratinocytes to proteins within the lamina densa (basement membrane proper) and sublamina densa regions of the epidermal basement membrane zone (see Ch. 28 ). Defects in protein components of these structures lead to various forms of EB (simplex, junctional and dystrophic; see Ch. 32 ) . The basement membranes of epithelia in other sites such as the eye, oral cavity, gastrointestinal tract, and genitourinary tract may also be affected in patients with EB. Additional clinical manifestations can result when the abnormal protein has important functions in extraepithelial tissues, such as the skeletal muscle for plectin (leading to EBS with muscular dystrophy).
Focal adhesions are structurally defined sites of linkage between the intracellular actin cytoskeleton and the extracellular matrix (ECM; see Fig. 28.3B ). Mutations in the gene encoding the focal adhesion protein kindlin-1 cause Kindler syndrome, an autosomal recessive disorder characterized by acral blistering and photosensitivity early in life, progressive poikiloderma and erosive periodontal disease (see Ch. 63 ). Kindlin-1 is found within basal keratinocytes along the cell surface that faces the basement membrane, and it has roles in keratinocyte adhesion, polarity, proliferation and motility. To date, Kindler syndrome represents the only skin fragility disorder known to be caused by disruption of linkage between actin microfilaments (rather than keratin intermediate filaments) and the ECM .
Transmembrane Transporter Defects
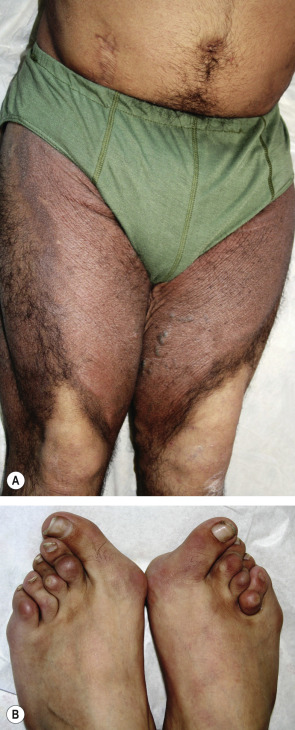
Abnormalities in transmembrane transporters underlie a variety of genodermatoses, ranging from occipital horn syndrome and Menkes disease (both due to mutations in the gene encoding the ATP7A copper transporter) to oculocutaneous albinism types 2 and 4 (due to defective transporters on lysosome-related organelles) to metabolic disorders such as acrodermatitis enteropathica, Hartnup disease, and hemochromatosis. This list also includes defects in a fatty acid transporter in ichthyosis prematurity syndrome and an intracellular nucleoside transporter in H syndrome ( Fig. 55.2 ) . Disorders of calcium pumps and ATP-binding cassette (ABC) transporters, which underscore the important roles of transmembrane transporters in cutaneous homeostasis, are discussed below.
Calcium pump defects
Darier disease and Hailey–Hailey disease are distinct autosomal dominant disorders that share the histologic feature of epidermal acantholysis leading to suprabasal cleft formation; on an ultrastructural level, this corresponds to disruption of the desmosome–keratin intermediate filament complex. The two conditions also have some clinical similarities, including intertriginous involvement, vesiculobullous lesions (relatively uncommon in Darier disease), nail changes, exacerbation with heat, and frequent superinfections. As a result of these overlapping features, it was initially postulated that Darier disease and Hailey–Hailey disease were allelic disorders caused by defects in a gene encoding a structural component of the epidermis. However, linkage studies mapped Darier disease to chromosome 12 and Hailey–Hailey disease to chromosome 3, and no candidate structural genes were found in these regions.
In 1999, pathogenetic mutations in the ATP2A2 gene, which encodes a sarcoplasmic/endoplasmic reticulum Ca 2+ ATPase, were identified in patients with Darier disease. Subsequently, mutations in the ATP2C1 gene on chromosome 3, which encodes a Ca 2+ ATPase localized to the Golgi apparatus, were shown to cause Hailey–Hailey disease. Determining the molecular bases of these genodermatoses thus demonstrated the pivotal role of calcium homeostasis in epidermal differentiation and cell–cell adhesion .
ATP-binding cassette transporter defects
Members of the ABC transporter superfamily bind and hydrolyze adenosine triphosphate (ATP) in order to transport various molecules across a cell membrane or into a vesicle. Well-known examples include the cystic fibrosis transmembrane conductance regulator (CFTR, ABCC7; the chloride ion channel that is defective in patients with cystic fibrosis) and the P glycoprotein (ABCB1; an important cellular mechanism of multidrug resistance). Several phenotypically diverse genodermatoses are also caused by mutations in ABC transporters (see Table 55.1 ) .
Because fragmentation and calcification of elastic fibers in the skin, eyes, and cardiovascular system are major features of pseudoxanthoma elasticum (PXE), the latter was initially thought to represent a heritable connective tissue disorder that (presumably) resulted from mutations in a gene encoding an extracellular matrix protein. Surprisingly, PXE was instead found to be caused by mutations in the ABCC6 gene, which encodes an organic acid transporter (based on in vitro studies) that is expressed almost exclusively in the liver and kidneys. Together with evidence that metabolites present in the sera of PXE patients interfere with the normal assembly of elastic fibers in vitro , this suggests that PXE is actually better characterized as a metabolic disorder with secondary connective tissue manifestations.
Biallelic mutations in ABCA12 , which encodes a transporter that secretes lipids into lamellar granules, cause both harlequin ichthyosis and a form of lamellar ichthyosis (see Fig. 56.2 ). Patients with harlequin ichthyosis tend to have nonsense mutations, whereas those with lamellar ichthyosis typically have missense mutations. This represents one of the most clearcut genotype–phenotype correlations observed to date for non-syndromic autosomal recessive ichthyoses. Defective formation of intercellular lipid layers, which are essential for epidermal barrier function, due to genetic defects in lipid metabolism also represents the pathomechanism of an expanding list of other ichthyosiform disorders (see Table 55.1 and Fig. 56.2 ) .
Nuclear Envelope Defects
“Nuclear envelopathies” represent a clinically heterogeneous group of inherited disorders caused by defects in structural components of the nuclear envelope (see Table 63.10 ). In addition to a variety of extracutaneous findings – e.g. skeletal dysplasia, muscular dystrophy, cardiomyopathy and neuropathy – dermatologic manifestations include: premature aging of the skin, e.g. in progeria and “atypical Werner syndrome” due to LMNA mutations; cutaneous fibrosis, e.g. in restrictive dermopathy and Buschke–Ollendorff syndrome/melorheostosis due to LMNA and LEMD3 mutations, respectively; and partial lipodystrophy, e.g. familial Dunnigan and “acquired” Barraquer–Simons types due to LMNA and LMNB2 mutations, respectively.
“Laminopathies” represent the largest subgroup of envelopathies and result from mutations in the genes that encode the lamin A/C and lamin B2 proteins of the nuclear membrane lamina, which lies just inside the inner nuclear membrane . The LEMD3 gene encodes MAN1, an inner nuclear membrane protein that associates with lamin A. In addition to “true” envelopathies, mutations in the ZMPSTE24 gene, which encodes a zinc metallopeptidase involved in the processing of prelamin A into mature lamin A, can result in restrictive dermopathy and mandibuloacral dysplasia characterized by lipodystrophy, premature aging and skeletal defects. The study of patients with envelopathies has provided insight into the importance of lamins and other nuclear envelope proteins to the structural integrity of the nucleus, chromatin organization, transcriptional regulation, control of differentiation, and mechanisms of aging .
Defects in Pyrin/NOD Family Members and Related Proteins
The molecular bases of several hereditary periodic fever syndromes and other autoinflammatory disorders have been found to be due to defects in proteins containing n ucleotide-binding o ligomerization d omains (NODs) and/or pyrin domains (see Table 45.2 ) . This has drawn attention to the NOD and pyrin families and related proteins (see Fig. 4.2 ), helping to uncover their importance in innate immune responses and acquired inflammatory disorders. Defects in such proteins lead to diverse cutaneous phenotypes, ranging from urticarial papules in cryopyrin-related disorders and erysipeloid erythema in familial Mediterranean fever to nodulocystic acne and pyoderma gangrenosum in PAPA syndrome to granulomatous dermatitis (together with arthritis and uveitis) in Blau syndrome (see Fig. 45.11C ). Upon uncovering NOD2 mutations as the defect in Blau syndrome, it was also established that this autosomal dominant disorder and “early-onset sarcoidosis” are identical on a genetic as well as phenotypic level (with the latter resulting from de novo NOD2 mutations), thus representing a single disease entity. However, different NOD2 variants have been implicated in Crohn disease and susceptibility to leprosy.
Defects in DNA Repair Genes, Tumor Suppressor Genes and Oncogenes
Hereditary cancer syndromes that affect the skin can be divided into groups based upon whether the associated malignancies are primarily cutaneous (e.g. basal cell nevus syndrome; see Table 55.6 ) or extracutaneous (see Table 55.7 ); individuals in the latter group may have either benign cutaneous neoplasms (e.g. Cowden syndrome) or non-neoplastic skin lesions (e.g. ataxia–telangiectasia) . To complement this clinical approach, molecular classification of such syndromes can help to explain relationships among disorders, understand the disrupted pathway or process, and suggest candidate genes for patients with similar phenotypes for which the underlying molecular defect has not yet been identified. In addition to characterization based on the specific pathomechanism (e.g. RAS–MAPK pathway activation ; see below), hereditary cancer syndromes can be separated into three general categories: (1) defective DNA repair and protection of genomic integrity (“caretaker” genes); (2) defective tumor suppressor (“gatekeeper”) genes; and (3) activated oncogenes.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


