Fetal cutaneous wounds in early gestation regenerate without the formation of a scar across mammalian species.
Myriad differences in the extrinsic and intrinsic components of scarless fetal and scarring adult wound healing have been identified.
Although the exact mechanism of fetal scarless wound regeneration remains unknown, recent advances in wound healing research hold promise for better understanding and recapitulating this phenomenon.
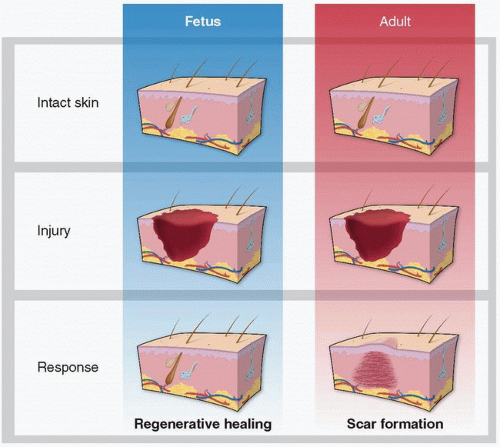
As discussed in other chapters, wound healing is a complex mechanism coordinated by multiple cell types involving numerous biologic pathways. It is highly evolved to protect against injury and infection. However, adult mammalian wound repair reproducibly results in the formation of a fibrotic scar. The resultant scar tissue quickly restores the epithelial barrier but is distinct from normal skin. Because of incomplete regeneration of the original tissue by a fibroproliferative response and an overproduction of poorly organized collagen, the resultant scar has a tensile strength that is less than 80% of its original form1 (see Chapter 6). Scar tissue is further characterized by a flattened epidermis and loss of dermal appendages, such as hair follicles and sebaceous glands (see Chapter 5).2 Humans are uniquely burdened with the ability to undergo pathologic scarring resulting in keloids or hypertrophic scars. These pathologic scars are characterized by an excessive deposition of collagen, sometimes extending beyond the original wound, resulting in a prominent scar often complicated by pain, itching, and/or devastating psychosocial consequences (see Chapters 9, 11, 19, and 24).3
However, the end result of wound healing depends on the developmental stage of the injured organism as well as the type of tissue damaged. A landmark manuscript in 1979 described that intrauterine wound healing in an early gestational human fetus does not result in scar formation.4 Numerous studies since then have demonstrated fetal wound regeneration in early to mid-gestational fetal skin in both human and other mammalian models.5,6,7 The transition from scarless wound regeneration to the postnatal scarring phenotype is dependent on gestational age and wound size, with larger wounds requiring an earlier gestational age to undergo regeneration.3 This occurs at about 24 weeks of gestation in humans and around embryonic day 18.5 (E19) in mice (term = day 21.5 or E22).8 Interestingly, the transition from regeneration to repair varies on the type of tissue. For example, myocardial regeneration occurs in neonatal mice until postnatal day 7.9 In addition, oral mucosal wounds heal with little to no scar formation at an accelerated rate, even in the adult.10,11,12
Scarless fetal wound healing was initially thought to occur because of the environmental conditions; however, studies have demonstrated that the intrauterine environment is neither necessary nor sufficient for regeneration.13 Instead, the capacity for regeneration is intrinsic to the tissue itself. Although the exact mechanisms underlying scarless fetal wound healing are yet unknown, understanding this privileged wound repair continues to be an area of investigatory interest with hopes of recapitulating this phenomenon in adult wound healing. Herein, we discuss the major differences in scarless mammalian cutaneous fetal wound healing as compared to scar-forming adult wound healing (Fig. 27-1).
Extrinsic Components of Fetal Wound Healing
To better understand the differences in wound healing capabilities in fetal and adult tissue, we classify contributions as extrinsic or intrinsic based on their relationship to the healing wound (Table 27-1). In terms of the differences between fetal and adult wound healing, extrinsic properties relate largely to the inflammatory response. The blunted inflammatory response observed in fetal wound healing tissue is understood to contribute to scarless wound healing. In this section we consider the cells that contribute to this inflammatory response, as well as the signaling molecules implicated in the process.
Inflammatory Cells
As in adult wound healing, circulating cells are recruited to the wound to initiate the inflammatory response. This sequence, beginning with neutrophils, followed by macrophages and mast cells, is maintained in fetal wound healing. It is in the magnitude and intensity of the inflammatory response that separates the privileged fetal wound from scar formation as seen in adult cutaneous wound repair.
Adapted from Hu MS, Maan ZN, Wu JC, et al. Tissue engineering and regenerative repair in wound healing. Ann Biomed Eng. 2014;42(7):1497-1507.
Neutrophils
Neutrophil recruitment is usually initiated by platelet degranulation at the wound bed during initial hemostasis after wounding. It is, therefore, not surprising that the diminished inflammatory response in fetal wound healing is partly attributed to differences in platelet activity. Platelet morphology is consistent across fetal and adult tissues, but fetal platelets have been shown to produce lower levels of transforming growth factor β1 (TGF-β1) and platelet-derived growth factor.14 They also demonstrate suboptimal aggregation in response to collagen prior to fetal transition from scarless healing to scar formation.15 Tumor necrosis factor α (TNF-α) and interleukin-1 (IL-1) are also released during platelet degranulation. Together these molecules act as chemoattractants, directing circulating neutrophils to wounded dermal tissue. TNF-α and IL-1 also play a role in upregulating neutrophil adhesion molecules, which have been found to be much less abundant among fetal neutrophils.16 Neutrophil migration to the fetal wound bed is limited by lower neutrophil-endothelial cell interactions secondary to attenuated adhesion molecule expression.17,18 The absence of subpopulations of proinflammatory and anti-inflammatory neutrophils further differentiates the early stages of the fetal wound healing response from that found in adults.19
Macrophages
Macrophage localization to the wound bed follows neutrophil infiltration.18 Persistent macrophage activity in wounds is thought to cause excess scar formation.20 Studies of embryonic and fetal murine models demonstrate that macrophages are not recruited to wounds prior to gestational day 14 (E14.5), except in cases of substantial tissue damage.21 Interestingly, fetal transition to scar formation occurs later at around E19 (term = E22).8 Despite the potential for macrophage recruitment during this window period between E14.5 and E19, their activation remains diminished because of noticeably lower TGF-β1 levels.22 This growth factor is in part responsible for the transition of circulating monocytes to activated macrophages. Conversely, TGF-β3 acts as a stop signal for terminal differentiation, and is at its peak concentration during the period of scarless fetal wound healing.23,24 This favorable ratio of TGF-β1:β3 contributes to the privileged scarless wound healing phenotype.
Mast Cells
Mast cells are the third and final population to localize to the wound environment. The mast cell response is approximately halved in healing fetal tissues, and those that are recruited to the wound bed were noted to be more immature and less able to undergo degranulation. Fetal mast cells also release lower levels of inflammatory mediators including histamine, TGF-β, TNF-α, and vascular endothelial growth factor (VEGF), reducing neutrophil chemotaxis and extravasation.25 Although mast cells were once thought to contribute to scar formation during fetal wound healing,25 subsequent work has shown that they are not required for healing of cutaneous full-thickness excisional wounds in adult mice.26
Inflammatory Modulators
Regulation of inflammatory processes occurs throughout all phases of wound healing via the action of cytokines, some of which have been discussed previously. As with almost any process within the body, a balance is maintained through opposing forces. The same is true for wound healing with various cytokines acting as either agonists or antagonists to an inflammatory response. As alluded to previously, the general principle that differentiates scarless fetal wound healing from normal wound healing is that the balance is shifted in favor of an anti-inflammatory response. Key inflammatory modulators in wound healing include members of the TGF-β family, ILs, and VEGF.
Transforming Growth Factor β
Three isoforms of TGF-β exist1,2,27 whose patterns of expression differ between adult and fetal wounds. Adult wounds demonstrate a greater quantity of TGF-β1 and TGF-β2, whereas TGF-β3 predominates in fetal wounds.28 The finding that fetal wounds are relatively hypoxic compared to adult wounds suggests a potential role for hypoxia-inducible factor 1α in preferential TGF-β3 regulation.29 In fact, elevated oxygen tensions have been shown to impede fetal fibroblasts.30 TGF-β1 plays a major role in adult wound inflammation, stimulating chemotaxis in neutrophils, monocytes, and fibroblasts. The positive feedback loop generated by these infiltrating cells releasing further TGF-β1 is thus inhibited in fetal wounds, where there is a relative dearth of this proinflammatory cytokine. As mentioned previously, the TGF-β3 dominating the fetal wound response acts as a stop signal for terminal differentiation of tissue. Fetal cells at the wound bed may be held at a more immature state secondary to TGF-β3, with their less fully developed inflammatory machinery implicated in scarless healing in the fetus.
Interleukins
Similar to the TGF-β cytokine family, subtypes of ILs play antagonistic roles in the inflammatory response to acute wounding. Levels of IL-6 and IL-8, known proinflammatory signaling molecules highly expressed during adult wound healing, are diminished in the fetal wound.18,31 IL-10, however, acts as an anti-inflammatory cytokine. Fetal wounds have been found to overexpress IL-10,32 whereas its absence in fetal IL-10 knockout mice predisposes to scar formation that would otherwise not occur.33 IL-10 inhibits proinflammatory expression of IL-1, IL-6, IL-8, and TNF-α34; it also inhibits inflammatory cell migration while allowing for normal collagen deposition and dermal architecture.32
Vascular Endothelial Growth Factor
VEGF is fundamental to angiogenesis in adult wound healing, but its upregulation has also been observed in association with keloid and hypertrophic scar development.35,36,37,38,39 VEGF expression in fetal wound healing has been noted in some studies to be relatively tempered, resulting in fetal wounds that are less vascular than their fibrotic counterparts.40 Our understanding of the exact role of VEGF in fetal wound healing is incomplete as results are not consistent across all studies. Colwell et al.41 noted significant VEGF upregulation in scarless wound repair occurring in E16 fetal mice compared to scarring repair in E18. However, despite this finding, there was little difference in terms of neovascularization based on histologic findings. Beyond stimulation of angiogenesis, VEGF does act in a proinflammatory manner by increasing vascular permeability and promoting inflammatory cell infiltration of the wound bed. Antibody-mediated neutralization of VEGF resulted in up to a 75% reduction in scar width.40 Taken together, these findings suggest that VEGF plays a multifactorial role in modulating wound healing, whether up- or downregulating scar-forming processes.
Only gold members can continue reading. Log In or Register to continue