Abstract
Inherited epidermolysis bullosa (EB) represents a group of genetic disorders characterized by mechanically fragile skin with a propensity to develop blisters and/or erosions. EB simplex, junctional EB, dystrophic EB and Kindler syndrome represent the four major types of EB, which differ in the ultrastructural site within which cutaneous blisters form – intraepidermal, intra-lamina lucida, sublamina densa and mixed, respectively. At least 39 distinct subtypes of EB have been described. The diagnosis is based on the clinical phenotype, ultrastructural and immunohistochemical findings, and molecular genotype. Many EB subtypes are associated with substantial morbidity and even mortality related to skin and mucosal involvement, extracutaneous complications, and cutaneous malignancies (primarily SCCs). Management of EB is currently focused on the prevention of mechanical trauma, wound care, and avoidance as well as treatment of infections and extracutaneous manifestations. Advances in our knowledge of the pathogenesis of this group of diseases may lead to the development of more effective gene-, protein- and cell-based therapies.
Keywords
epidermolysis bullosa simplex, junctional epidermolysis bullosa, dystrophic epidermolysis bullosa, Kindler syndrome, basement membrane, skin fragility, epidermolysis bullosa, mechanobullous disease
- ▪
Epidermolysis bullosa (EB) encompasses multiple clinically distinctive disorders that share three major features: genetic transmission, mechanical fragility of the skin, and blister formation
- ▪
There are four major forms of inherited EB – EB simplex, junctional EB, dystrophic EB, and Kindler syndrome – which differ in the ultrastructural site within which cutaneous blisters form
- ▪
EB may be diagnosed by immunofluorescence antigenic mapping, transmission electron microscopy, or genetic analysis
- ▪
Any epithelial-lined or epithelial-covered organ is at potential risk for involvement in the more severe forms of inherited EB
- ▪
In the absence of specific therapy, management is primarily focused on prevention of blisters, wound care, and treatment of extracutaneous complications
Introduction
Inherited epidermolysis bullosa (EB), the prototypic mechanobullous disease, is characterized by the development of blisters following seemingly minor or insignificant trauma or traction to the skin . It currently encompasses four major forms – EB simplex, junctional EB, dystrophic EB, and Kindler syndrome – and at least 40 distinctive clinical phenotypes ( Table 32.1 ). Inherited EB can result from mutations within the genes for any of at least 19 structural proteins: keratins 5 and 14; the subunits of laminin 332 (formerly laminin 5); types VII and XVII collagens; plectin; α 6 β 4 integrin; α 3 integrin subunit; bullous pemphigoid antigen 1; kindlin-1 (fermitin family homolog-1); exophilin 5; transglutaminase 5; kelch-like protein 24; and the desmosomal components plakophilin-1, plakoglobin and desmoplakin. Although most of these diseases are rare, research into their underlying pathophysiologic bases has led to major advances in our understanding of the cell and molecular biology of keratins, other keratinocyte-associated structural proteins, collagens, and the cutaneous extracellular matrix (ECM). Study of EB has also helped to elucidate mechanisms of epithelial cell adhesion, migration, and differentiation and to highlight the role of the basement membrane zone in health and disease. The creation of in vitro and animal models of EB has enabled principles of gene therapy to be tested and refined, with development of novel approaches for the treatment of EB patients.
History
Epidermolysis bullosa was first described in 1870 by von Hebra under the name “erblichen pemphigus” . Its current name, “epidermolysis bullosa hereditaria”, was coined by Koebner in 1886. Hallopeau recognized the distinct clinical features of simplex and dystrophic forms of EB in 1898. Junctional EB was first identified in 1935 by Herlitz and termed “EB letalis”. Precise characterization of these three major EB types via the use of transmission electron microscopy was first achieved by Pearson in 1962 . In the ensuing years, additional EB phenotypes were described. Monoclonal antibody studies provided the first suggestion that specific protein defects underlie individual types and subtypes of this disease . In 1986, the National EB Registry was established in the US by the National Institutes of Health, facilitating epidemiologic, clinical, and laboratory characterization of each major EB type and subtype . In 1991, Bonifas et al. utilized linkage analysis to demonstrate the molecular basis for EB simplex. Subsequent work by others has established the precise molecular basis for most of the EB subtypes recognized to date.
Epidemiology
Based on National EB Registry data , the estimated overall prevalence and incidence of EB in the US are 11.1 per 1 million population and 19.6 per 1 million live births, respectively. Carrier frequencies have also been reported . The approximate prevalences and incidences, respectively, of the major types of EB are as follows: EB simplex, 6.0 and 7.9; junctional EB, 0.5 and 2.7; dominant dystrophic EB, 1.5 and 2.1; and recessive dystrophic EB, 1.4 and 3.0.
Pathogenesis
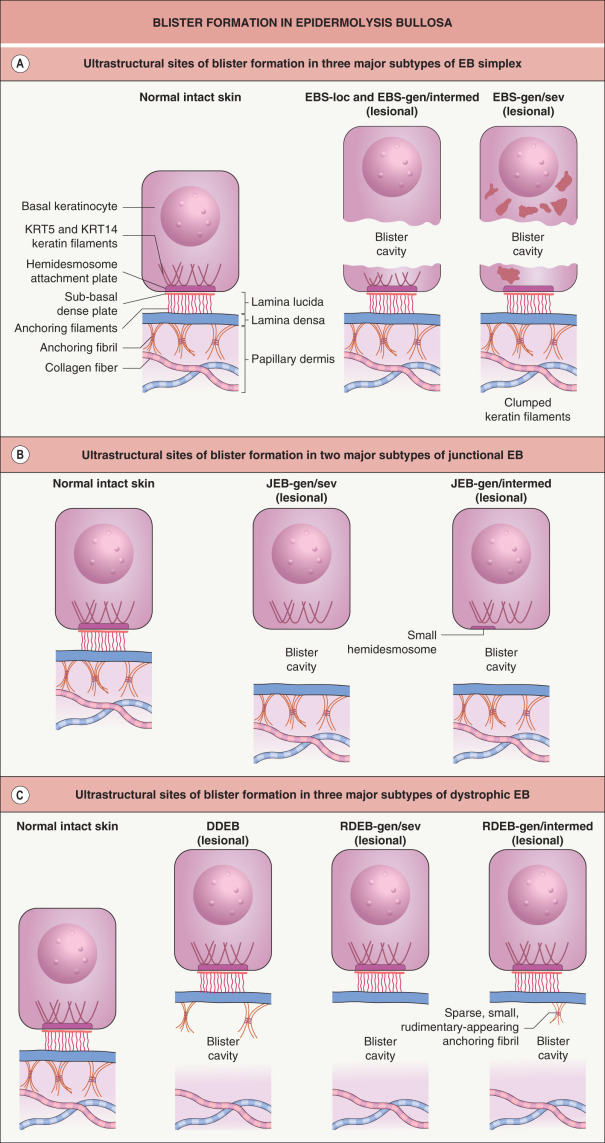
EB is caused by mutations within genes that encode structural proteins that reside within the epidermis (EB simplex), dermal–epidermal junction (junctional EB), or uppermost papillary dermis (dystrophic EB). The site within which each of these proteins resides determines the ultrastructural location where the blisters arise ( Table 32.1 & Fig. 32.1 ).
EB Simplex
The most common forms of EB simplex (EBS) are transmitted in an autosomal dominant manner. There are two main subgroups of EBS, suprabasal and basal, which differ in the ultrastructural level of the intraepidermal blistering (see Table 32.1 ). The vast majority of EBS cases are in the basal group, most often resulting from a dominant-negative mutation (see Ch. 54 ) within the keratin 5 ( KRT5 ) or 14 ( KRT14 ) genes, expression of which is primarily within the basal layer of the epidermis . The clinical severity and other phenotypic features of EBS are closely associated with the genotype. For example, mutations in the helix initiation and termination motifs of KRT5 and KRT14 (see Fig. 56.5 ) lead to the generalized severe subtype of EBS (EBS-gen/sev, previously known as EBS-Dowling–Meara), whereas the specific phenotype of EBS with mottled pigmentation almost always arises from a particular missense mutation in the V1 domain of KRT5. Dominant stabilizing mutations in the kelch-like protein 24 (KLHL24) ubiquitin ligase lead to EBS via increased ubiquitination and degradation of KRT14 . An autosomal recessive form of EBS due to mutations in the gene encoding plectin is associated with muscular dystrophy, which is not surprising considering that plectin is expressed in skeletal muscle as well as in the hemidesmosomes of basilar keratinocytes. Other EBS patients with plectin or α 6 β 4 integrin deficiency present with pyloric atresia, while rare autosomal recessive variants of basal EBS caused by mutations in the bullous pemphigoid antigen 1 or exophilin 5 genes have also been reported. In addition, suprabasal forms of EBS result from mutations in the genes encoding transglutaminase 5 and the desmosomal proteins plakophilin-1, plakoglobin, and desmoplakin (see Table 32.1 and Fig. 56.8 ).
Junctional EB
Junctional EB (JEB) is almost always transmitted in an autosomal recessive manner (see Table 32.1 ). The generalized severe subtype of JEB (JEB-gen/sev, previously known as JEB-Herlitz) typically results from homozygous or compound heterozygous truncating mutations within a gene encoding one of the three subunits of laminin 332, a key component of the lamina lucida of the dermal–epidermal junction (see Ch. 28 ) . In laryngo-onycho-cutaneous syndrome, the underlying mutations affect only the a isoform of the laminin α3 subunit. The milder generalized intermediate form of JEB (JEB-gen/intermed, previously known as JEB-non-Herlitz and generalized atrophic benign EB) results from mutations within the genes for either a subunit of laminin 332 or type XVII collagen. JEB with pyloric atresia, which is more common than EBS with pyloric atresia, is also caused by mutations within either of the two genes that encode the subunits of α 6 β 4 integrin. A newly recognized form of JEB associated with respiratory and renal involvement results from mutations in the integrin α 3 chain.
Dystrophic EB
Dystrophic EB (DEB) is transmitted in either an autosomal dominant or autosomal recessive manner and is caused by mutations in the type VII collagen gene. Dominant DEB (DDEB) results from dominant-negative mutations, typically a missense mutation that leads to substitution of another amino acid for a glycine within the triple-helical domain of this collagen. Although the resultant protein is structurally abnormal, immunohistochemical staining of the dermal–epidermal junction is usually indistinguishable from that of normal skin.
Recessive DEB (RDEB) is usually due to compound heterozygous mutations within the type VII collagen gene . Premature stop codons, which result in truncated proteins, are characteristic of the generalized severe subtype (RDEB-gen/sev, previously known as RDEB-Hallopeau–Siemens). Consistent with the severity of these mutations, anchoring fibrils are undetectable or extremely sparse and poorly formed in skin biopsy specimens, and immunohistochemical staining with antibodies against the major epitopes of the type VII collagen molecule is absent or barely detectable. Milder forms of generalized RDEB are associated with less severe biallelic mutations in the type VII collagen gene.
Recently, a single nucleotide polymorphism in the matrix metalloproteinase 1 gene promoter has been identified as a disease modifier in RDEB . It is likely that additional modifying genes account for some of the inter- and intra-familial phenotypic variability observed in this and other forms of EB. There is also evidence for secondary effects from loss of protein expression in EB. For example, loss of collagen VII in RDEB fibroblasts leads to alterations in dermal matrix proteins, metalloproteinases, and transforming growth factor β (TGF-β), which are thought to further influence keratinocyte adhesion and dermal–epidermal integrity .
Some patients with RDEB retain the amino terminal non-collagenous domain (NC1) of type VII collagen, and this specific fragment may contribute to an increased susceptibility to develop squamous cell carcinomas (SCCs) . In a Ras-driven model of tumorigenesis, RDEB keratinocytes that contained no type VII collagen did not form tumors in mice, whereas RDEB keratinocytes that produced the NC1 domain of type VII collagen were tumorigenic. Additional studies have pointed to the fibronectin-like sequences within NC1 as key to promoting tumor cell invasion. However, SCCs can also develop in RDEB patients who do not express the NC1 domain .
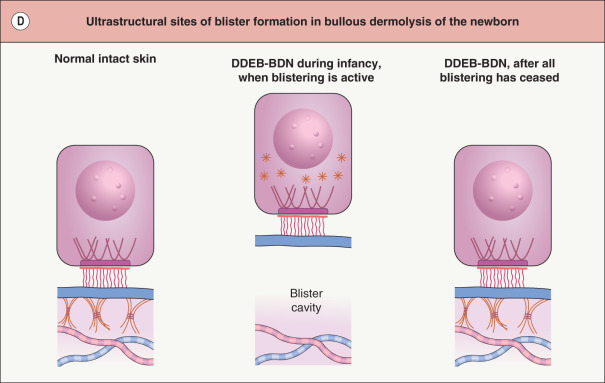
In a rare form of DEB referred to as bullous dermolysis of the newborn, inheritance is usually dominant and blistering is typically confined to the first 1–2 years of life . Clinical expression coincides with a time during which type VII collagen is present primarily within basilar keratinocytes in these patients’ skin, rather than along the dermal–epidermal junction (see Fig. 32.1D ). This suggests that there may be a temporary disruption in the transport of this protein from the keratinocyte cytoplasm to the underlying ECM.
Clinical Features
Cutaneous Findings
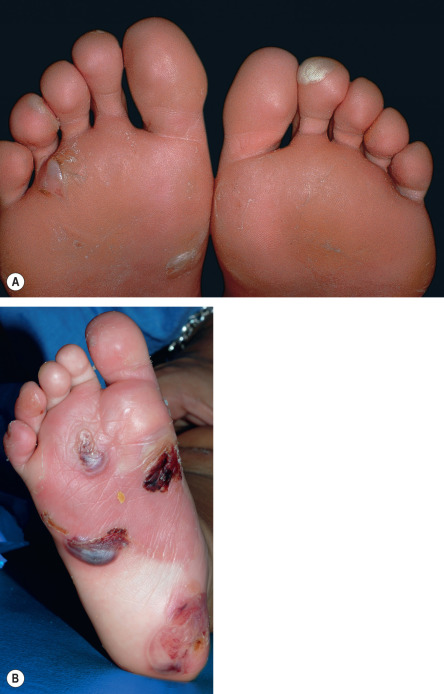
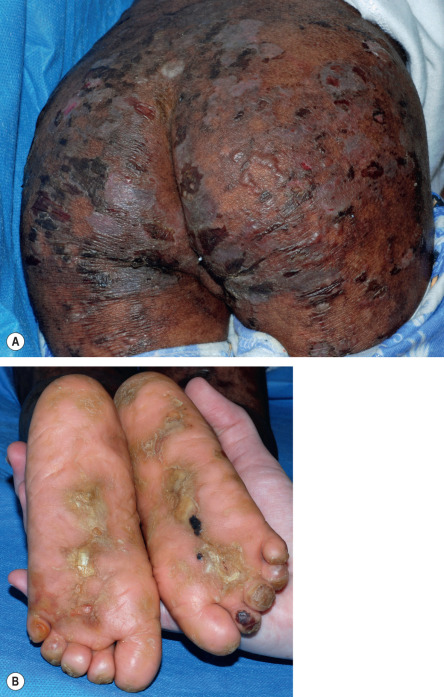
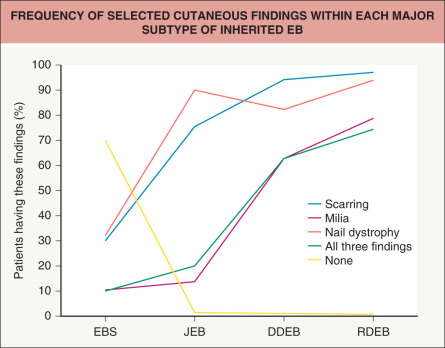
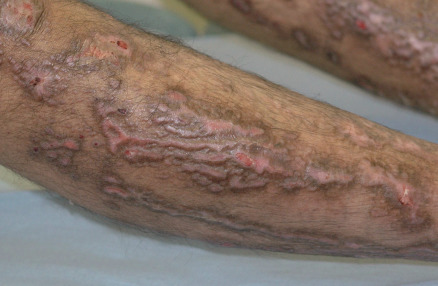
All forms of inherited EB are characterized by mechanically fragile skin, erosions, and (with rare exceptions) macroscopic blisters ( Figs 32.2 & 32.3 ). Scarring is almost always atrophic and can occur in any subtype of EB ( Fig. 32.4 ), including localized EBS. However, scarring is most frequent in the subtypes that are characterized clinically by generalized disease activity and ultrastructurally by disruption of the basement membrane, particularly the lamina densa ( Fig. 32.5 ). Whereas scarring is estimated to occur in only 15% of patients with localized EBS, it is present in essentially every patient with RDEB . Other cutaneous findings that have similar variations in frequency across the major types and subtypes of EB include dystrophic or absent nails (see Fig. 32.4 ), milia, and scarring alopecia of the scalp.
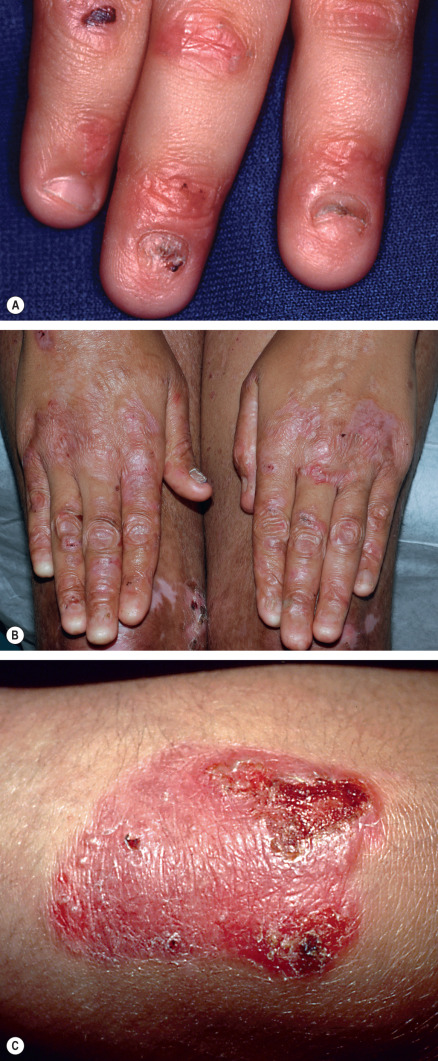
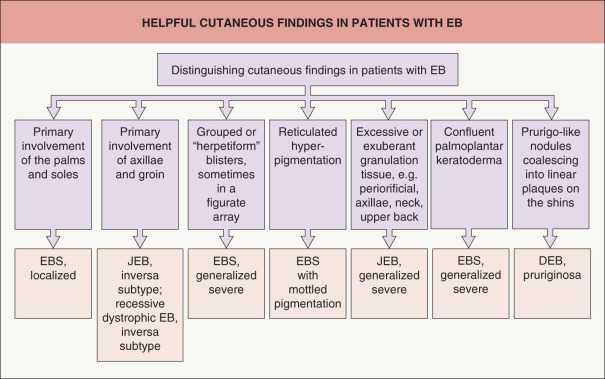
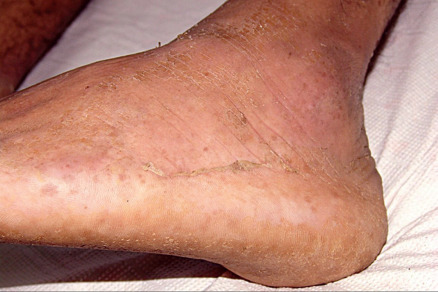
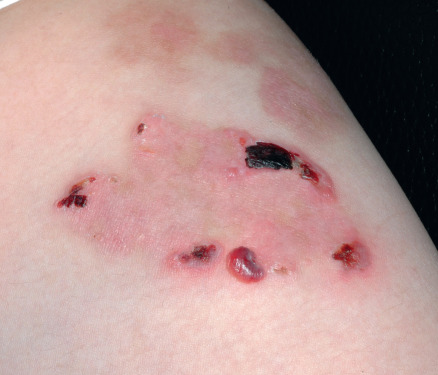
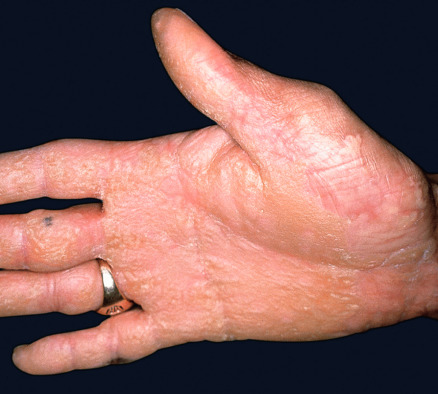
Some cutaneous findings have diagnostic implications ( Fig. 32.6 ) . Reticulated hyperpigmented macules distinguish a rare subtype of EB simplex termed EBS with mottled pigmentation . Superficial peeling of the skin, often in the absence of overt blistering, is seen in EBS superficialis (EBSS) and acral peeling skin syndrome ( Fig. 32.7 ) . Grouped (“herpetiform”) blisters, often in an arcuate ( Fig. 32.8 ) or polycyclic array, are highly characteristic of EBS-gen/sev , which is also associated with the gradual development of a diffuse palmoplantar keratoderma ( Fig. 32.9 ). Migratory circinate erythema with vesiculation of the advancing edge has been described in patients with EBS caused by a frameshift mutation that leads to an elongated KRT5 protein. Excessive or exuberant granulation tissue, usually in a symmetric distribution involving the periorificial areas, skin folds, upper back, nape of the neck, and periungual areas, is typical of JEB-gen/sev ( Fig. 32.10 ). Extremely pruritic papules coalescing in a linear arrangement on the lower extremities are characteristic of DEB pruriginosa ( Fig. 32.11 ).
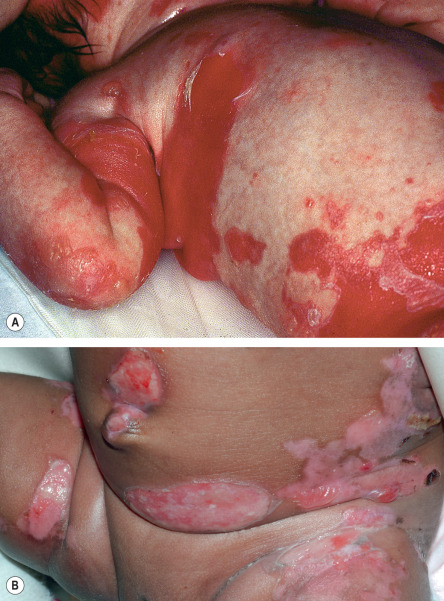
Distribution of skin disease activity is also useful in the subclassification of EB , although the pattern of involvement tends to be less distinctive in infants than in adults. Patients with the “ inversa ” subtypes of JEB and RDEB experience severe disease activity primarily in intertriginous areas such as the axillae and inguinal creases. In contrast, patients with localized JEB have involvement mainly in acral sites, and those with pretibial DEB have lesions almost exclusively on the shins. RDEB centripetalis is a rare subtype that initially features acral blistering, followed by slow progression of disease activity toward the trunk over the years.
Extracutaneous Findings
The molecular defects that affect the skin in patients with EB may also lead to manifestations in other tissues with an epithelial lining or surface , including the eye, oral cavity, and gastrointestinal, genitourinary, and respiratory tracts. Major extracutaneous complications of EB are summarized in Table 32.2 . Although exceptions exist, extracutaneous involvement occurs most frequently in RDEB and JEB, and it can result in blisters, erosions, ulcers, and scarring. Rare subtypes of JEB and EBS present at birth with pyloric atresia as well as skin fragility and blistering . In other forms of EB, extracutaneous disease may become apparent as early as the first few months of life. Repeated blistering of the external eye can result in neovascularization and blindness . Chronic involvement of the esophagus leads to scarring, stricture formation and, rarely, even complete obstruction . Involvement of the small intestine presents with chronic malabsorption, whereas disease activity within the large intestine tends to produce constipation and anal fissures or strictures. Recurrent genitourinary tract blistering may result in urethral or ureterovesical strictures; if persistent, the latter may eventuate in ureteric reflux and hydronephrosis. Tracheolaryngeal blistering and associated soft tissue edema, seen most often in infants and young children with JEB-gen/sev , may lead to potentially fatal acute airway obstruction . JEB with respiratory and renal involvement is a rare subtype associated with severe interstitial lung disease.
| MAJOR EXTRACUTANEOUS COMPLICATIONS OF EPIDERMOLYSIS BULLOSA | ||
|---|---|---|
| Complication | EB subtype(s) most commonly affected | |
| ≥50% of patients | <50% of patients | |
| Eyes | ||
| Corneal blisters, ulcers and scarring | RDEB-gen/sev | JEB-gen/sev > RDEB-gen/intermed, RDEB-inv, JEB-gen/intermed |
| Ectropion formation | JEB-gen/sev; Kindler | |
| Oral cavity and upper airway (excluding blisters) | ||
| Microstomia | RDEB-gen/sev | JEB-gen/sev, RDEB-inv |
| Enamel hypoplasia | JEB (all subtypes) | |
| Excessive caries and premature loss of teeth | RDEB gen/sev, JEB-gen/sev | |
| Tracheolaryngeal stenosis | JEB-gen/sev | JEB-gen/intermed |
| Gastrointestinal tract | ||
| Esophageal strictures | RDEB-gen/sev, RDEB-inv | RDEB-gen/intermed, Kindler > JEB-gen/sev |
| Pyloric atresia | JEB-PA, EBS-PA | |
| Malnutrition/failure to thrive | RDEB-gen/sev, JEB-gen/sev | JEB-gen/intermed |
| Severe constipation | RDEB-gen/sev | JEB-gen/sev, EBS-gen/sev |
| GERD | RDEB | JEB, EBS-gen/sev |
| Colitis | Kindler, RDEB-gen/sev | |
| Genitourinary tract | ||
| Urethral meatal stenosis | RDEB-gen/sev, JEB-gen/sev, Kindler | |
| Chronic renal failure * | RDEB-gen/sev | |
| Hydroureter and hydronephrosis | JEB-PA, JEB-gen/sev | |
| Nephrotic syndrome | JEB-resp/renal | JEB-gen/sev |
| Heart | ||
| Dilated cardiomyopathy | RDEB-gen/sev > JEB, RDEB-gen/intermed | |
| Musculoskeletal system | ||
| Pseudosyndactyly | RDEB-gen/sev | RDEB-gen/intermed, Kindler |
| Osteoporosis or osteopenia | RDEB-gen/sev | RDEB-gen/sev, JEB-gen/sev |
| Muscular dystrophy | EBS-MD | |
| Bone marrow | ||
| Severe multifactorial anemia | RDEB-gen/sev, JEB-gen/sev | |
Dental enamel hypoplasia, which occurs in all forms of JEB , is associated with pitting of the surfaces of primary and permanent teeth. If untreated, affected individuals lose teeth during childhood due to excessive caries . Severe caries and resultant tooth loss also occur in RDEB-gen/sev , likely resulting from impaired clearance of food from the mouth and poor dental hygiene in the setting of intraoral injury and scarring, ankyloglossia, and microstomia.
Pseudosyndactyly (“mitten” deformities) of the hands and feet primarily affects patients with RDEB, especially RDEB-gen/sev , although it occasionally occurs to a lesser degree in DDEB and JEB ( Fig. 32.12 ). Initially presenting as proximal web formation between adjacent digits, the digits may eventually become totally fused and encased by scar tissue. Lack of mobility leads to bone resorption and muscular atrophy, and hand function is severely compromised.
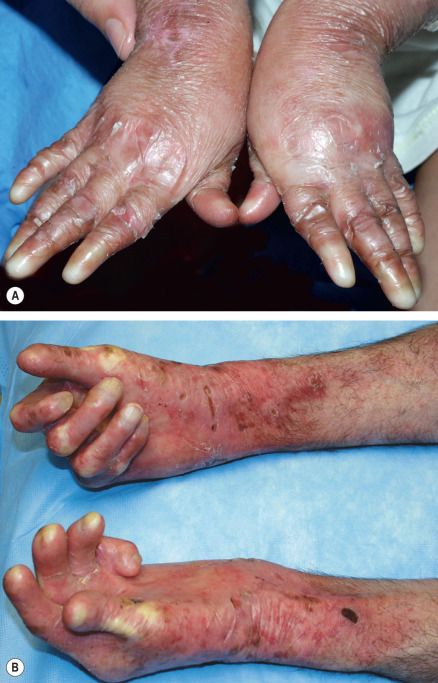
Osteoporosis, detectable by dual-emission X-ray absorptiometry (DEXA) scanning, is common in RDEB-gen/sev and JEB-gen/sev . Radiographs may demonstrate vertebral crush fractures in severe cases. EBS due to plectin deficiency is associated with mild to severe muscular dystrophy. Although the muscle disease presents during infancy in some patients, weakness often develops insidiously during later childhood or even early adulthood in those who are less severely affected.
Chronic renal failure occasionally develops in patients with severe forms of EB, most notably RDEB-gen/sev , which is associated with a ~10% risk of death from renal failure by age 35 years . Renal disease may result from untreated outflow obstruction, glomerulonephritis, secondary systemic amyloidosis or IgA nephropathy. Nephrotic syndrome associated with altered expression of laminin isoforms in renal basement membranes has been reported in an infant with JEB-gen/sev and it occurs congenitally as a result of integrin α 3 mutations in JEB with respiratory and renal involvement. A small subset of patients with severe forms of EB, especially RDEB-gen/sev , develop potentially fatal dilated cardiomyopathy . Although as yet unproven, selenium or carnitine deficiency may be a contributing factor.
Although it was a common occurrence several decades ago, potentially lethal bacterial sepsis is now relatively rare in inherited EB , presumably due to improved wound care and the availability and judicious use of broad-spectrum antibiotics. When sepsis does occur in EB, it tends to affect infants with generalized severe disease . In contrast, failure to thrive is still common among infants with JEB-gen/sev and may lead to death.
Cutaneous Malignancies
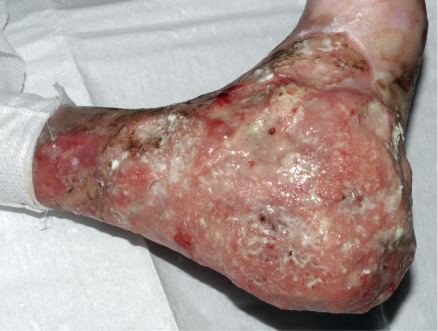
A major complication, usually of RDEB, is the development of multiple cutaneous SCCs . These tumors most often arise in chronic non-healing wounds or hyperkeratotic lesions ( Fig. 32.13 ). Histologically, they are usually well differentiated. However, the borders of the lesions are often indistinct and they are difficult to completely excise, with a tendency to recur locally. In addition, SCCs in EB patients frequently metastasize and are strikingly unresponsive to chemotherapy or radiotherapy. Indeed, they represent the leading cause of death in EB at or after mid-adolescence, with death from an SCC of cutaneous origin occurring in most patients within 5 years of the diagnosis of their first SCC . These tumors occur primarily in RDEB, especially RDEB-gen/sev, although SCCs can also develop in adults with generalized JEB. The cumulative risk of at least one SCC in patients with RDEB-gen/sev is 7.5% by age 20 years but rises to 68%, 80%, and 90% by ages 35, 45, and 55 years, respectively . In contrast, the risk of SCC in other forms of RDEB is <25% by age 45 years .