Although chiefly of cosmetic significance, disorders of pigmentation are among the most conspicuous and thus can have profound psychosocial implications for pediatric patients. The most important pigments in skin are melanin, reduced and oxygenated hemoglobin, and carotene. Melanin is a pigment produced by melanocytes, specialized dendritic cells derived from the neural crest that migrate to the basal layer of the epidermis during embryogenesis. Melanocytes synthesize and package melanin within discrete membrane-bound organelles called melanosomes , which are then transferred via melanocytic dendrites to surrounding keratinocytes of epidermis and hair follicles; on average, there is one melanocyte to every 36 surrounding keratinocytes. Variations in skin color among different individuals reflect the number and size of mature melanosomes, not the number of melanocytes.
Four stages of melanosome maturation have been described and can be distinguished by ultrastructural examination:
- 1.
Membrane vesicles that contain no visible pigment (stage I or premelanosomes)
- 2.
More elongated vesicles with an ordered internal membrane but no pigment (stage II melanosome)
- 3.
The presence of melanin on ordered internal fibers (stage III melanosome)
- 4.
Structures so full of melanin that the luminal structures cannot be seen (mature or stage IV melanosomes).
Darkly pigmented individuals have more numerous, larger, singly dispersed melanosomes, whereas individuals with light pigmentation have fewer, smaller melanosomes that are aggregated into complexes and are more rapidly degraded. The presence of melanin in the epidermis helps protect against ultraviolet (UV) radiation and associated cutaneous damage, including pigmented nevi, actinic damage, and cutaneous neoplasia. Red hair color, usually associated with an inability to tan, increases the risk of developing melanoma fourfold and has been associated with polymorphisms in the melanocortin receptor 1 (MCR1).
Melanin exists in two forms in human skin: brown-black eumelanin and yellow-red pheomelanin. Melanin biosynthesis is primarily regulated by tyrosinase, a copper-dependent enzyme that allows the initial conversion of tyrosine to dihydroxyphenylalanine (DOPA). Eumelanin synthesis involves increased levels of tyrosinase activity and additional melanogenic enzymes such as tyrosinase-related protein (TRYP)-1 and TRYP-2/dopachrome tautomerase, both regulators of distal steps in the pathway to melanin and/or stabilizers of tyrosinase. Pheomelanin synthesis, however, involves the addition of a cysteinyl group that accounts for the yellow-red color and is associated with reduced tyrosinase activity and absence of TRYP-1, TRYP-2, and a protein called pink-eyed dilution (P) protein.
The ratio of eumelanin to pheomelanin, as well as the total content of melanin, is higher in skin types V–VI (the darkest skin colors) than in skin types I and II (the lightest skin colors, most prone to burning with UV light exposure). Pheomelanin levels tend to be greatest in individuals with bright red hair, whereas eumelanin is the predominant pigment in individuals with brown or black hair.
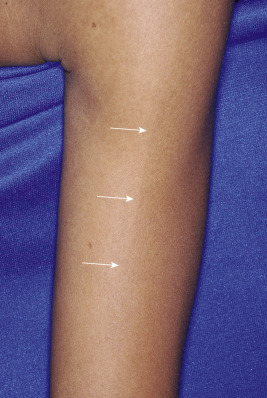
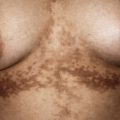
In all races the dorsal and extensor surfaces are relatively hyperpigmented, and the ventral surfaces are less pigmented. This is most evident in races with darker skin (African-Americans, Hispanics, and Asians). The separation of the dorsal and ventral pigmentation is most conspicuous on the extremities (Voigt–Futcher lines) ( Fig. 11-1 ). This differentiation of dorsal and ventral pigmentation is present from infancy and persists throughout adulthood. Approximately 75% of African-Americans and 10% of whites have at least one line of pigmentary demarcation.
Disorders of Abnormal Pigmentation
Disorders of decreased pigmentation may be classified as:
- 1.
Genetic or developmentally controlled disorders, in which pigmentation tends to be abnormal from birth or early infancy
- 2.
Disorders associated with depigmentation or loss of previously existing melanin.
Genetic disorders of decreased pigmentation include tuberous sclerosis, piebaldism, Waardenburg syndrome (WS), and albinism. Acquired disorders of decreased pigmentation include vitiligo, postinflammatory hypopigmentation, pityriasis alba, and tinea versicolor. Hypopigmentary disorders may be further divided into patterned and unpatterned groups. Patterned forms of decreased pigmentation include pityriasis alba, cutaneous T-cell lymphoma, tinea versicolor, postinflammatory hypopigmentation, leprosy, pinta, tuberous sclerosis, pigmentary mosaicism, vitiligo, piebaldism, and the Waardenburg and Vogt–Koyanagi syndromes. Unpatterned decreases in pigmentation may be seen in albinism, phenylketonuria, and the silvery hair syndromes.
Vitiligo
Vitiligo, an acquired form of patterned loss of pigmentation, is a polygenic, multifactorial disorder that involves at least 16 susceptibility genes, encoding a variety of proteins involved in regulation of the immune system and tyrosinase, the principal vitiligo autoimmune antigen. Melanocytes are destroyed by antigen-specific cytotoxic T cells, resulting in patchy depigmentation. Both lesional and nonlesional skin show up regulation of markers of heightened innate immunity, although only lesional skin shows suppression or absence of melanocyte-specific genes. Autoantibodies that can destroy melanocytes have been detected in serum samples of patients with vitiligo, further emphasizing that alterations are not limited to lesional skin.
Vitiligo affects approximately 1% of the population. Although rarely congenital, its onset in about half of affected patients is before the age of 20 years and in one-quarter before 8 years of age. The disorder has a prevalence of 7% to 12% among first-degree relatives, 6% among siblings, and 23% among monozygotic twins, but a recent study suggested a family history of vitiligo in approximately 30% of patients. Relative to postpubertal onset (after 12 years of age), prepubertal onset is associated with a greater likelihood of a family history of vitiligo and a personal history of atopic dermatitis. Autoimmune disorders are seen with significantly increased incidence in immediate family members of affected individuals, most commonly vitiligo itself, but other autoimmune disorders (particularly hypothyroidism and alopecia areata) occasionally occur in pediatric patients with vitiligo. Thyroid autoimmune antibodies have been described in 11% of patients.
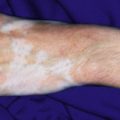
The location, size, and shape of individual lesions vary considerably, yet the overall picture is characteristic. Lesions usually appear as partially or completely depigmented ivory-white macules or patches, usually with well-defined, sometimes hyperpigmented, convex borders ( Figs. 11-2 through 11-7 ). They tend to have an oval or linear contour and range in size from several millimeters to large patches. Rarely, extensive or near-total depigmentation of the body (universal or total vitiligo) occurs (see Fig. 11-4 ). Although usually considered to be a bilateral disorder, vitiligo may be asymmetric; segmental vitiligo, in which the depigmentation is confined to a localized, usually unilateral area, occurs more often in children than in adults (see Fig. 11-5 ). In 75% of affected individuals the first lesions occur as depigmented spots on exposed areas such as the dorsal surfaces of the hands, face, and neck. Other sites of predilection include the body folds (the axillae and groin), body orifices (the eyes, nostrils, mouth, navel, areolae, genitalia, and perianal regions (see Figs. 11-6 and 11-7 ), and areas over bony prominences such as the elbows, knees, knuckles, and shins (see Fig. 11-3 ). Approximately 12% of patients show white hairs (leukotrichia or poliosis) ( Fig. 11-8 ). Vitiligo has been divided into several subtypes based on the distribution of lesions. In descending order of incidence in pediatric patients, these include generalized, focal, segmental, acrofacial, mucosal, and universal. Patients with vitiligo, especially those with prepubertal onset, commonly show halo nevi, pigmented nevi surrounded by a zone of depigmentation ( Fig. 11-9 ) (see Chapter 9 , Fig. 9-26 ). The discovery of a halo nevus and particularly multiple halo nevi should prompt the search for vitiligo elsewhere.
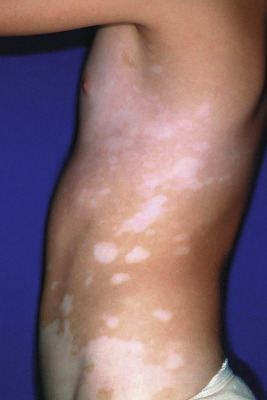
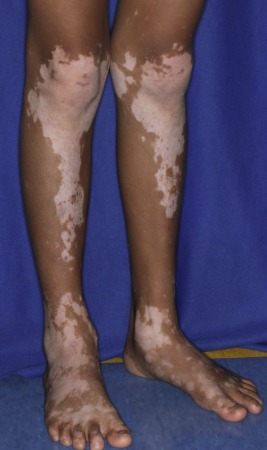
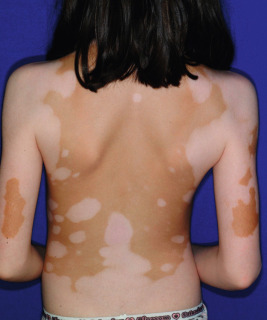
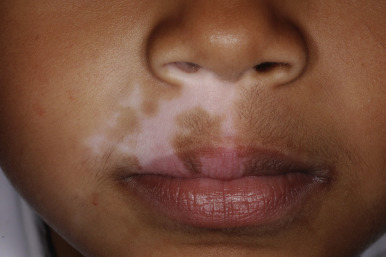
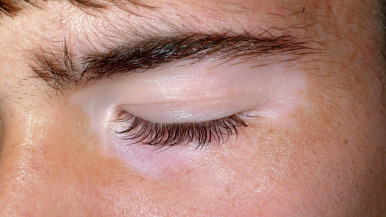
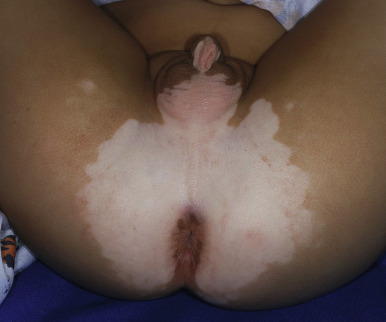
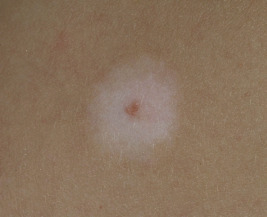
The Koebner phenomenon (development of a lesion after trauma) has been described in approximately 15% of affected children with vitiligo, particularly related to sunburn. A recent study showed that skin friction induces melanocyte detachment in persons with vitiligo but not in individuals with normal skin, further emphasizing the role of trauma in triggering new lesions. Other individuals associate the onset of vitiligo with periods of severe physical or emotional trauma.
Ordinarily the diagnosis of vitiligo is not difficult, especially when there is symmetric depigmentation about the eyes, nostrils, mouth, nipples, umbilicus, or genitalia. In fair-skinned individuals it may be difficult to differentiate areas of vitiligo from the adjacent normal skin. In such cases examination under Wood light in a darkened room may help delineate a contrast between normal and depigmented skin. When the diagnosis is in doubt, the distribution of lesions, the age at onset, the presence of a convex hyperpigmented border, and the characteristic sites of predilection may help establish the correct diagnosis.
Lesions of postinflammatory hypopigmentation are hypopigmented, not depigmented, and patients usually provide a history of previous localized inflammation. However, it is not uncommon to see residual depigmentation in severe atopic dermatitis, especially in darker skinned patients and involving the wrists, hands, ankles, and feet. Nevus depigmentosus tends to be a well-defined, usually hypopigmented patch that may be present at birth or appear during infancy as normal pigmentation increases but is subsequently stable (see Fig. 11-22 ). Pityriasis alba is a hypopigmentary disorder and may be further differentiated by its common distribution on the face, upper arms, neck, and shoulders and its occasional fine adherent scale (see Chapter 3 , Fig. 3-33 ). Lesions of tinea versicolor may be differentiated by their discrete or confluent small, round hypopigmented macules; their fine scales; and their typical distribution on the trunk, neck, upper arms, or particularly in pediatric patients, the face (see Chapter 17 , Figs. 17-33 and 17-34 ). The demonstration of hyphae on microscopic examination of epidermal scrapings is confirmatory (see Fig. 17-36 ). The diagnosis of cutaneous T-cell lymphoma of the hypopigmented type should be considered in adolescents with more extensive hypopigmented macules resembling pityriasis alba (see Chapter 10 , Fig. 10-21 ).
The presence of a white forelock and the pattern of depigmentation suggest a diagnosis of piebaldism or WS (see Figs. 11-17 and 11-19 ). Most individuals with WS show characteristic facial features. The diagnosis of albinism (see Oculocutaneous Albinism section) may be established by its presence at birth and by the facts that normal eye color is retained in vitiligo (but not in albinism); in addition, hair on glabrous skin in the patient with vitiligo, in contrast to that in the patient with albinism, often retains most of its pigment (see Fig. 11-11 ). Adolescents with GM3 synthase deficiency progressively develop depigmented patches as well as acral lentigines. The hypopigmented macules of tuberous sclerosis (see Fig. 11-21, A ) usually lack the characteristic milk-white appearance of lesions of vitiligo, are present at birth or during the first years of life, do not change with age, and have a normal number of melanocytes (with reduction in size of melanosomes and melanin granules within them) in contrast to the absence or decrease in number of melanocytes in patients with vitiligo.
The course of vitiligo is variable. Long periods of quiescence may be interrupted by periods of extension or partial improvement. Complete spontaneous repigmentation is very rare, and in one study, more than 50% repigmentation occurred in only 2.4% of patients over 6 months. At least partial repigmentation is more likely in children with lesions of recent onset and during the summer months because of increased exposure to UV light. Loss of pigmentation in lesions that have at least partially repigmented is common in temperate climates during the winter months. The repigmentation process proceeds slowly, although children tend to respond with more permanent and complete repigmentation than adults. Repigmentation most commonly appears as small, freckle-like spots of repigmentation, reflecting the migration of melanocytes from the hair follicle ( Fig. 11-10 ). As such, the chance of repigmentation in a site is greater if pigmentation of regional hairs is retained. Diffuse repigmentation of lesions or repigmentation from the margins has also been described. The preferential tendency to repigment the face and neck versus other body sites has been attributed to the high density of hair follicles at these sites, as well as exposure to UV light. In contrast, sites lacking or poor in hair follicles, such as the dorsal surfaces of the fingers, hands, feet, and the volar aspect of the wrists, do not respond as well as other areas.

Quality-of-life studies have shown that children with vitiligo have impaired social development as young adults, stressing the importance of intervention. Risk factors associated with the highest risk for quality-of-life impairment are age (15 to 17 years), location (face and legs), and greater extent of involvement. In one study, anxiety was observed in 42% of caregivers of children with vitiligo, even higher than that in caregivers of children with atopic dermatitis or psoriasis, and was correlated with poor quality-of-life scores in the children. Patients with vitiligo and their families can find support through the National Vitiligo Foundation ( www.vitiligofoundation.org or www.nvfi.org ).
Full repigmentation is challenging, especially in children with more extensive involvement and when vitiligo involves more recalcitrant areas. At least partial repigmentation can often be accomplished by the twice-daily application of mid-strength to potent topical corticosteroids or topical calcineurin inhibitors (tacrolimus ointment, pimecrolimus cream). The skin of the head and neck responds best to both of these treatment modalities. Overall, 40% to 90% of pediatric patients show a response to these treatments, although moderate to high potency steroids can theoretically be associated with systemic absorption, especially if applied over large body-surface areas or on the head and neck continuously. Application of a topical calcineurin inhibitor, especially for facial vitiligo, eliminates the risk of cutaneous atrophy and ocular toxicity carried by application of topical corticosteroids. However, hyperpigmentation in sun-exposed areas has been described after use of tacrolimus ointment. Although topical anti-inflammatory therapy has been standard, one study described good to excellent repigmentation in 65% of 400 children treated with minipulses of oral methylprednisolone on 2 consecutive days weekly and fluticasone ointment twice daily. Topical application of vitamin D 3 analogues (calcipotriene, calcipotriol) has also been used but has the potential to be more irritating.
The repigmentation of lesional skin can be stimulated most effectively by exposure to UV light, generally in combination with topically applied anti-inflammatory medications or vitamin D 3 analogues. Avoidance of burning with phototherapy is important, because cutaneous burning can lead to further depigmentation via the Koebner phenomenon. Most commonly narrow-band (nb) UVB is utilized, because it has been shown to be as effective as psoralen and UVA (PUVA) therapy, which is now rarely used in children because of its toxicity. nbUVB phototherapy is largely reserved for older pediatric patients who are highly motivated and completely informed about their chances for improvement with these therapies. Treatment is traditionally 2 to 3 times weekly, beginning at a relatively low dose and increasing by about 20% each treatment until slight erythema is reached. Should there not be a good response within 6 months, nbUVB can be stopped. The 308-nm monochromatic excimer laser (in the UVB range) is a painless therapy for more localized lesions. The best responses to excimer laser are at sites that also respond best to nbUVB, with the dorsal aspect of the hands and feet, genital area, and suprapubic area the most difficult sites to repigment. In one study of chronic stable vitiligo, more than 50% of patients showed more than 75% repigmentation. Responses to the excimer laser may be improved by concurrently using anti-inflammatory therapy. Treatment with the combination of topical tacalcitol (vitamin D derivative) and excimer laser for 12 sessions (over 12 weeks) was significantly more effective than the excimer laser treatments alone. The use of antioxidants, particularly pseudocatalase, has been based on the demonstration of decreased enzymatic and nonenzymatic oxidants in the skin of patients with vitiligo. Although in one retrospective uncontrolled study, twice-daily full-body application of pseudocatalase cream coupled with daily low-dose nbUVB stopped vitiligo progression and led to more than 75% repigmentation in 93% of treated children, most experience with pseudocatalase cream has been disappointing. Oral antioxidants for vitiligo are under investigation.
Surgical modalities are based on the autologous grafting of nonlesional epidermis or cultured melanocytes from healthy skin sites to depigmented areas that have been deepithelialized by ablative procedures. Chinese cupping has recently been shown to be a technique to induce blisters for capturing donor melanocytes. Grafting has been demonstrated to lead to at least 75% repigmentation in 30% to 90% of patients and is most successful for more localized lesions. Although minigrafting with or without UV light exposure has shown success, especially in patients with facial grafts and with segmental and limited subtypes, these approaches are time-consuming, costly, and can result in recurrence of vitiligo (including at the donor site), scarring, infection, and keloids in at-risk patients; grafting should only be considered for stable vitiligo in selected adolescents at sites that are resistant to medical treatment.
When treatment is unsatisfactory, lesions can be hidden by the use of camouflage therapy, which has been shown to improve the quality of life in children with vitiligo. Camouflage can be achieved most effectively with cosmetics (e.g., Cover FX, Dermablend, or Covermark), but aniline dye stains, such as Vitadye (Elder) and quick-tan preparations have also been used.
In those few recalcitrant cases in which vitiligo has progressed to such an extent that more than 50% of the body is involved (particularly in those persons in whom only a few islands of normal skin remain), an attempt at depigmentation with 20% monobenzyl ether of hydroquinone (Benoquin) may be considered. Such patients should be reminded that the depigmentation is permanent, requiring lifelong vigilant use of sun protection. Owing to the permanence of depigmentation therapy, this treatment is not generally offered to preadolescent patients.
Vogt–Koyanagi–Harada Syndrome
Vogt–Koyanagi–Harada syndrome is a rare autoimmune disorder characterized by bilateral granulomatous uveitis, alopecia, vitiligo, poliosis, dysacousia (in which certain sounds produce discomfort), deafness, and sometimes meningeal irritation or encephalitic symptoms. Usually seen in adults in the third and fourth decades of life, the disorder also occurs in children and adolescents.
The bilateral uveitis occurs in all patients and generally takes a year or more to clear. The uveitis is often accompanied by choroiditis and optic neuritis. As the uveitis begins to subside, poliosis (in 80% to 90%), usually bilateral vitiligo (in 50% to 60%), alopecia (in 50%), and temporary auditory impairment develop. A prodromal febrile episode with lymphocytosis, encephalitic or meningeal symptoms, and increased pressure of the cerebrospinal fluid may precede the bilateral uveitis. The poliosis may be limited to the eyebrows and eyelashes or may also involve the scalp and body hair. The pigmentary changes, which generally appear 3 weeks to 3 months after the onset of the uveitis, tend to be permanent. Although most patients show some recovery of visual acuity, the majority of children and adolescents have a residual visual defect related to the development of cataracts, glaucoma, choroidal neovascularization, and subretinal fibrosis.
Early and aggressive systemic corticosteroids are the primary intervention, but refractory cases may respond to cyclosporine, methotrexate, or tumor necrosis factor (TNF) inhibitors.
Alezzandrini Syndrome
Alezzandrini syndrome is a rare disorder of unknown origin primarily seen in adolescents and young adults. Possibly related to Vogt–Koyanagi–Harada syndrome, it is characterized by unilateral degenerative retinitis with visual impairment followed after an interval of months or years by bilateral deafness and unilateral vitiligo and poliosis, which appear on the side of the retinitis.
Oculocutaneous Albinism
Albinism is a group of inherited disorders of melanin synthesis manifested by a congenital decrease of pigmentation of the skin, hair, and eyes. Although some classifications include nonsyndromic and syndromic (e.g., silvery hair and Hermansky–Pudlak syndromes) forms, the pigmentary changes are very different among these disorders. Most albinism is oculocutaneous, but affected individuals may have ocular albinism, usually an X-linked recessive form caused by mutation in OA1/GPR143, with the abnormal pigmentation limited to the eye. An oculocerebral syndrome with hypopigmentation (Cross–McKusick–Breen syndrome) is characterized by oculocutaneous albinism (OCA), microphthalmos, spasticity, and mental retardation. Although albinism associated with immunodeficiency is primarily seen in the silvery hair syndromes (especially Chédiak–Higashi and Griscelli type 2), immunodeficiency is a feature of OCA associated with short stature (owing to mutations in LAMTOR2 ) and in Hermansky–Pudlak syndrome (HPS) types 2 and 9, all related to the requirement for secretion of lysosomes and cytosolic granules for cytotoxic T- and natural killer cell function, antigen presentation to T cells, and neutrophil antimicrobial activity.
Nonsyndromic Oculocutaneous Albinism
Nonsyndromic oculocutaneous albinism (OCA) encompasses seven subtypes ( Table 11-1 ) with decreased or absent melanin biosynthesis in the melanocytes of the skin, hair follicles, and eyes.
| Type | Percentage of Patients Worldwide | Mutation | Function of Affected Gene | Comments |
|---|---|---|---|---|
| OCA1A | 50% | TYR (absence) = tyrosinase negative | Critical enzyme in melanin formation | 1:40,000; most severe cutaneous and ocular defects; highest risk of skin cancer; most common type in Caucasians |
| OCA1B | TYR (decreased) | Critical enzyme in melanin formation | Subtypes: yellow mutant (yellow hair); platinum (metallic tinge); minimal pigment (only eyes darken) | |
| OCA1-TS | TYR (mutation site functions at higher temperatures) | Temperature-sensitive | Melanin at cooler sites (arms, legs) | |
| OCA2 | 30% | P protein | Melanosome biogenesis and normal processing and transport of TYR and TRYP1 | 1:36,000 (whites); TYR-positive; includes Brown albinism (1:3900–1:10,000; most common form in patients of African origin; more pigment with advancing age) |
| OCA3 (Rufous) | 3% | TRYP1 | Catalyzes oxidation of 5, 6-dihydroxyindole-2-carboxylic acid monomers into melanin and stabilizes TYR so it can leave endoplasmic reticulum for incorporation into melanosomes | 1:8500 Africans; reddish-bronze color to skin and hair |
| OCA4 | 17% | MATP/SLC45A2 | Membrane transporter in melanosomes; mutations misroute TYR | Rare (whites); 1:, 000 Japanese (27% of OCA in Japan); resembles OCA2 |
| OCA5 | Unknown (4q24) | – | One Pakistani family | |
| OCA6 | SLC24A5 | Solute carrier protein involved in melanosome maturation and melanin biosynthesis | Heterogeneous extent of pigmentation | |
| OCA7 | c10orf11 | Melanocyte differentiation | Rare |
OCA affects 1 in 17,000 persons in the United States. The highest prevalence (as high as 1% of the population) occurs in the indigenous Cuna tribe on the San Blas Islands off the coast of Panama. Affected Cuna children have been called moon children because they have marked photosensitivity and photophobia and prefer to go outdoors only at night. In some African tribes, the frequency is 1:1500. OCA is characterized by varying degrees of unpatterned reduction of pigment in the skin and hair, translucent irides, hypopigmented ocular fundi, and an associated nystagmus. Melanocytes and melanosomes are present in the affected skin and hair in normal numbers but fail to produce normal amounts of melanin. Regardless of subtype, affected individuals require vigorous sun protection of the skin and eyes and are at risk of adverse psychosocial effects because of the cosmetic aspects of albinism, especially in children from darker-skinned backgrounds. In addition to the stigma and potential social isolation, affected individuals in Africa have been maimed or killed because of the myths associated with albinism (contagion, body parts with magical and medicinal powers, intercourse with an affected woman will cure human immunodeficiency virus [HIV] infection).
In the past, albinism was divided into tyrosinase-negative and tyrosinase-positive forms based on the ability (tyrosinase-positive) or inability (tyrosinase-negative) of plucked hair to become pigmented in the presence of tyrosine or DOPA. Tyrosinase-negative albinism, now called type I albinism ( OCA1 ), results from absence (OCA1A) or partial reduction of the activity (OCA1B) of tyrosinase, the critical enzyme in melanin formation (see Table 11-1 ). The underlying genetic bases for most forms of tyrosinase-positive albinism are also known. Type II albinism (OCA2) results from the absence of P protein, OCA3 from absence of TRYP1, and OCA4 from mutations in membrane-associated transporter protein (MATP). Types OCA5–7 have been only been described in one to a few families (see Table 11-1 ).
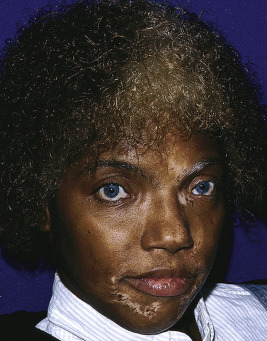
Individuals with OCA1A are unable to produce melanin at all and show white skin, white hair, and blue irides regardless of familial skin coloration. Hair may show a slight yellow tint with advancing age because of denaturation of hair keratins. Similarly, ocular abnormalities are most severe with OCA1A. Eye findings include photophobia (with squinting), nystagmus (which typically develops at 6 to 8 weeks of age), strabismus, and decreased visual acuity; patients with OCA1A are often legally blind. The optic fibers are misrouted, resulting in monocular vision, which is usually not altered by surgical correction of the nystagmus or strabismus. Although neurologic development is otherwise generally normal, an increased risk of attention-deficit/hyperactivity disorder (ADHD) has also been described. Actinic damage (cutaneous atrophy, telangiectasia and wrinkling, actinic cheilitis, actinic keratoses) and malignant skin tumors (especially squamous cell carcinoma and nodular basal cell carcinoma but also melanoma) are almost always seen in affected young adults but may present during childhood if the skin and eyes are not protected.
Patients with OCA1B have been divided into different phenotypic subgroups (see Table 11-1 ) that occur because of differences in degree of tyrosinase activity and the localization within the TYR gene of the mutation. In the yellow mutant form, the hair turns yellow in the first few years and a golden blond to light brown by the end of the second decade. Patients with platinum OCA develop small amounts of pigment with a metallic tinge in late childhood. Those with minimal-pigment OCA show darkening of the eyes with time, but the skin remains without pigmentation. Individuals with temperature-sensitive OCA1B are born with white skin and hair and blue eyes. Usually during the second decade of life, however, areas with lower temperature (especially hair at acral sites on the upper and lower extremities) are able to produce melanin, because the tyrosinase activity is only inactivated above 35 °C. This interesting phenotype is shared with that of the Siamese cat, a breed that also results from temperature-sensitive tyrosinase activity.
The OCA2 type of albinism, which includes tyrosinase-positive albinism and Brown OCA, is the most common form and is usually the type that occurs in African-American individuals ( Fig. 11-11 ). The phenotype may vary from a slight to moderate decrease in pigmentation of the skin, hair, and eyes. With time, however, dark lentigines and pigmented nevi usually develop at sun-exposed sites ( Fig. 11-12 ). These individuals can also have problems with their eyes and an increased risk of cutaneous malignancy, but significantly less than that seen in individuals with tyrosinase-negative albinism. Although the degree of pigment dilution in affected individuals is variable, the diagnosis is usually easily established in those who have striking pigment loss or relative pigment dilution when compared with unaffected siblings or parents. Some patients with OCA2 have red hair, which has been shown to result from concomitant mutations in the melanocortin 1 receptor. OCA2 has also been described in approximately 1% of patients with Angelman syndrome or Prader–Willi syndrome, disorders that result from deletion of the long arm of chromosome 15, the site of the P gene. Prader–Willi syndrome results from deletion of the paternal chromosome at 15q and is characterized by hyperphagia with obesity, hypogonadism, and mental retardation. In contrast, Angelman syndrome results from deletion of the maternal chromosome at 15q and is characterized by microcephaly, severe mental retardation, ataxia, and inappropriate laughter. Pigment dilution occurs when both copies of the P gene are mutated or deleted. Interestingly, duplication of the 15q chromosomal region has been associated with generalized skin hyperpigmentation.
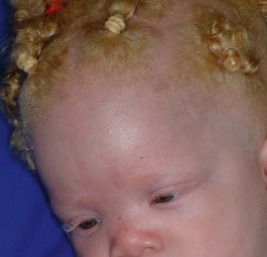
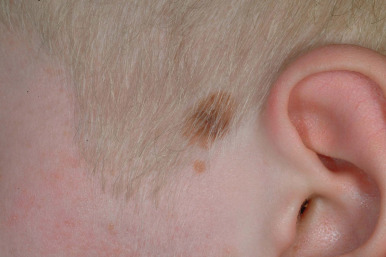
Rufous OCA or OCA3 presents as “ginger” red hair, a reddish-bronze color of skin, and blue or sometimes brown irides. This form may be underreported, because the decrease in pigmentation is slight and may be undetectable in lighter skinned patients. OCA4 is now considered one of the most common forms in Japan, and affected individuals resemble patients with OCA2.
Patients with albinism should be monitored by an ophthalmologist in addition to the dermatologist. Glasses may help the poor vision, and contact lenses and tinted glasses may ameliorate the photophobia. Nystagmus may be helped by surgery of the eye muscles or contact lenses; eye patching may be needed for the strabismus. High-contrast written material, large-type textbooks and computers that can enlarge text are all helpful for patients with poor visual acuity. Early actinic changes, keratoses, basal cell tumors, and particularly squamous cell carcinomas are common; the risk of melanoma (often amelanotic) is also increased, even in children and adolescents. Thus individuals with cutaneous albinism must learn to avoid sunlight exposure, to wear sunglasses, and to use protective clothing and sunscreen preparations on exposed surfaces. The National Organization for Albinism and Hypopigmentation (NOAH) is a national support group for patients and their families ( www.albinism.org ).
Hermansky–Pudlak Syndrome
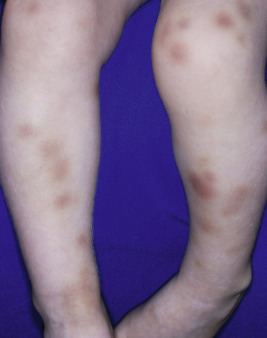
Hermansky–Pudlak syndrome (HPS) is a group of at least 9 autosomal recessive disorders (HPS1–9) characterized by pigment dilution, a hemorrhagic diathesis secondary to a platelet storage pool defect, and ceroid-lipofuscin depositions within the reticuloendothelial system, oral and intestinal mucosae, lung, and urine. HPS is a disorder of biogenesis of melanosomes and other lysosome-related organelles including platelet dense granules ( Table 11-2 ), and all of the mutations found are in genes encoding components of protein complexes (e.g., BLOC-1, BLOC-2, BLOC-3, and AP-3 ) that regulate vesicle trafficking in these organelle systems. HPS is most commonly seen in Hispanics from Puerto Rico (1:1800 to 1:400 persons; HPS-1 and sometimes HPS-3), in persons of Dutch origin, and in East Indians from Madras. The platelet defect in patients with HPS does not produce a severe problem in children. Its expression, however, can be aggravated by ingestion of aspirin and other prostaglandin blockers. Special precautions and sometimes platelet transfusions must be given to avoid excessive bleeding after minor trauma or dental surgery.
The diffuse pigmentary features of the skin and eyes of individuals with HPS include pigmentary dilution of the skin and often the irides with hair that has a peculiar sheen, although not as silvery as in Chédiak–Higashi syndrome (CHS), another syndrome of lysosome-related organelles. The degree of generalized pigment loss is quite variable in intensity, ranging from white skin to brown and light to brown eyes. Ocular pigmentation generally correlates with cutaneous pigmentation. Ocular findings include nystagmus, photophobia, and decreased visual acuity. Extensive ecchymoses are a common clinical manifestation ( Fig. 11-13 ). The bleeding diathesis also commonly manifests as epistaxis and menometrorrhagia. Patients with both HPS and systemic lupus erythematosus have been described. The life-threatening complications of HPS, other than the bleeding diathesis, have been described in certain subtypes and are unusual in most affected children. These include granulomatous colitis (including two patients with cutaneous granulomatous disease that resembled metastatic Crohn disease), progressive pulmonary fibrosis, and less commonly, cardiomyopathy and renal failure. Immunodeficiency is a features of HPS-2 and the newest described form, HPS-9, which to date has not been associated with a bleeding diathesis. The life expectancy is 30 to 50 years of age. Glasses or contact lenses can help to correct the refractive errors. The bleeding from skin wounds may be stopped with thrombin-soaked Gelfoam, and desmopressin (DDAVP) has been administered for tooth extraction and other invasive procedures. Transfusions of platelets or erythrocytes are occasionally required. The pulmonary fibrosis and enterocolitis are most commonly seen in HPS-1 and are often recalcitrant to therapy; infliximab is sometimes helpful. Gene testing is available for mutations of HPS1 and HSP3 .
Phenylketonuria
Phenylketonuria results from deficiency in phenylalanine hydroxylase, the enzyme that converts phenylalanine to tyrosine. Although rarely an issue because of widespread perinatal Guthrie testing, untreated patients with phenylketonuria may develop generalized hypopigmentation of the hair, skin, and/or eyes in comparison with family members, related to the deficiency of tyrosine, the substrate for melanin. Neurologic features predominate (mental retardation, seizures, hyperreflexia), but patients may also show dermatitis and rarely, focal morphea-like skin lesions (see Chapter 22 ). Treatment is by avoidance of dietary phenylalanine.
Silvery Hair Syndromes
Three syndromes, CHS, Griscelli syndrome (GS), and Elejalde syndrome (probably a subset of GS), are autosomal recessive disorders characterized by an early silvery sheen to the hair, relative pigmentary dilution of skin with a grayish coloration, and in some patients ocular hypopigmentation.
Chédiak–Higashi Syndrome
Patients with Chédiak–Higashi syndrome (CHS) usually have a characteristic silvery sheen to the hair and skin, with a skin color that may appear lighter than that of other family members. In affected individuals of family backgrounds of darker skin, however, the skin of acral, sun-exposed areas (ears, nose) may become intensely hyperpigmented ( Fig. 11-14 ) or show only speckled hypopigmentation. Decreased iris pigmentation results in an increased red reflex and photophobia. Strabismus and nystagmus are common, but visual acuity is usually normal. Inflammation and ulceration of the oral mucosa, especially of the gingivae, have been described.
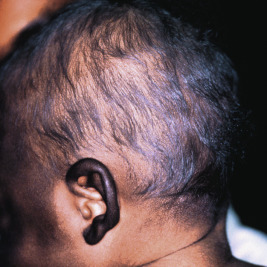
The immunodeficiency of patients with CHS leads to infectious episodes. These episodes are associated with fever and predominantly involve the skin, lungs, and upper respiratory tract. The most common organisms found are Staphylococcus aureus, Streptococcus pyogenes and pneumococcus. The skin infections are primarily pyodermas, but infections with these organisms that result in deeper ulcerations resembling pyoderma gangrenosum have been reported.
Approximately 50% to 85% of patients with CHS undergo an “accelerated” lymphohistiocytic phase during the first decade of life, characterized by widespread visceral tissue infiltration with lymphoid and histiocytic cells that are sometimes atypical in appearance. Hepatosplenomegaly, lymphadenopathy, pancytopenia, jaundice, a leukemia-like gingivitis, and pseudomembranous sloughing of the buccal mucosa are associated features. The thrombocytopenia, platelet dysfunction, and depletion of coagulation factors may lead to petechiae, bruising, and gingival bleeding. Granulocytopenia and anemia are found in 90% of patients during the accelerated phase. Neurologic manifestations may range from seizures to cranial nerve palsy to loss of consciousness. Viral infection, particularly from Epstein–Barr virus (EBV) infection, has been implicated in causing the accelerated lymphohistiocytic phase. The pigmentary changes that help to distinguish the CHS-related accelerated phase must be distinguished from autoimmune hemophagic syndromes and familial hemophagocytic lymphohistiocytosis (genetic defects in the cytolytic granule-dependent exocytosis pathway such as perforin). Neutropenia is common, and neutrophils are deficient in chemotactic and bactericidal capability. Selective deficiency of natural killer (NK) cells is characteristic. These immune abnormalities have been thought to cause the increased susceptibility to infections and the lymphohistiocytic phase.
Patients who survive into early adulthood may develop progressive neurologic deterioration, particularly with clumsiness, abnormal gait, paresthesias, and dysesthesias. Peripheral and cranial neuropathies and occasionally a form of spinocerebellar degeneration may occur.
CHS results from biallelic mutations in LYST, a lysosomal transport protein that regulates the fusion of primary lysosome-like structures. The skin pigmentary disorder has been attributed to the inability of melanosomal fusion and transfer to keratinocytes, leading to giant melanosomes within melanocytes. Giant granules are found in circulating leukocytes, melanocytes of skin and hair, renal tubular epithelial cells, central nervous system (CNS) neurons, and other tissues. In the hair shaft, these giant melanosomes are smaller than those of GS and regularly spaced ( Fig. 11-15, A ). The giant granules within phagocytic cells of affected children cannot discharge their lysosomal and peroxidative enzymes into phagocytic vacuoles. Of note, patients with leukemia may show granules that resemble those of CHS in leukocytes.


The mean age of death for patients with CHS without immune reconstitution is 6 years of age. Fatality usually results from overwhelming infection or hemorrhage during the accelerated phase. Approximately 10% to 15% of affected patients have a milder clinical phenotype and survive into adulthood but tend to develop the progressive neurologic dysfunction. For all of the silvery hair syndromes, early bone marrow or stem-cell transplantation is the treatment of choice for patients with a human leukocyte antigen (HLA) match. Bone marrow transplantation reverses the immunodeficiency and prevents the often-fatal accelerated phase but has no effect on pigmentation or on neurologic deterioration. Otherwise, management of the disorder is largely supportive. Antibiotics help to control the recurrent infections, and immunoglobulin or immunosuppressive agents have been administered in an attempt to control the lymphohistiocytic or hemophagocytic phases. Splenectomy has been advocated in patients with the accelerated phase unresponsive to other forms of therapy.
Griscelli Syndrome
Three subsets of patients with Griscelli syndrome (GS) have been described based on clinical manifestations and underlying gene defects. Two of these subsets are caused by mutations in genes close to each other on chromosome 15q21, myosin Va (type 1) and RAB27A (type 2). Myosin Va encodes a protein that binds organelles such as melanosomes to actin. RAB27A is a guanosine triphosphate (GTP)-binding protein involved in the movement of melanosomes. Melanocytes are unable to transfer melanosomes to epidermal cells, and ultrastructural examination of skin biopsies reveals accumulation of melanosomes in melanocytes but few in surrounding keratinocytes.
Patients who have uncontrolled activation of T lymphocytes and macrophages (hemophagocytic syndrome) and immune deficits (especially reduction in T-cell cytotoxicity and cytolytic granule exocytosis) have mutations in RAB27A , whereas those patients with neurologic problems and without immune abnormalities or hemophagocytosis tend to have myosin Va mutations. Elejalde syndrome (neuroectodermal melanolysosomal disease), characterized by similar abnormal pigmentation in the skin and hair and severe neurologic dysfunction (seizures, severe hypotonia, ocular abnormalities, and mental retardation) but no immunodeficiency or hemophagocytosis is now considered a subset of GS type 1. Rare patients with neurologic abnormalities with or without hemophagocytosis have shown a RAB27A mutation. Type 3 GS shows a phenotype restricted to the pigmentary defects and results either from mutation in the gene that encodes melanophilin ( MLPH ) or from a deletion in the F-exon of myosin Va.
Patients with GS, especially GS type 2, may be difficult to distinguish clinically from patients with CHS, because the silver-gray hair and skin color ( Fig. 11-16 ), recurrent episodes of fever with or without infection, increasing hepatosplenomegaly owing to lymphohistiocytic infiltration, and progressive neurologic deterioration may be part of the clinical spectrum of both disorders. In contrast to CHS, the lymphohistiocytic infiltration tends to occur in the first year of life. Blood smears show pancytopenia, but in contrast to the patients with CHS, no leukocyte inclusions. Microscopic examination of hair shows clumping of pigment in the hair shaft similar to that of CHS but with larger, more irregularly spaced macromelanosomes (see Fig. 11-15, B ).
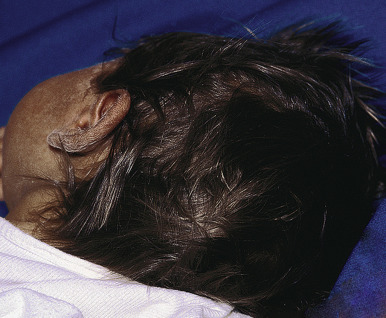
Intervention is similar to that of CHS, primarily through hematopoietic stem-cell transplantation. Given the lack of success of transplantation in patients with mutations in myosin Va, restriction of transplantation to patients with RAB27A mutations has been suggested. Early transplantation can prevent complications including the neurologic sequelae.
Piebaldism
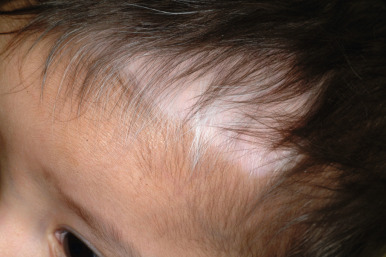
Piebaldism is an autosomal dominant disorder characterized by congenital patterned areas of depigmentation, including a white lock of hair above the forehead (the white forelock) in most affected individuals. The disorder usually results from mutations in the KIT protooncogene, which encodes a cell-surface receptor for the stem-cell/mast cell growth factor ; deletions in SNAI2/SLUG (encoding snail homolog of 2), a transcription factor, have also been described. The clinical manifestations of piebaldism may be explained by the resultant defective migration of melanoblasts from the neural crest to the ventral midline and a defect in the differentiation of melanoblasts to melanocytes. The distinctive patterns of hypopigmentation or depigmentation usually persist unchanged throughout life, but affected individuals with progressive depigmentation, response to UV light and partial repigmentation, or forelock regression during infancy have occasionally been described. The white forelock, with a depigmented triangular patch of the scalp and forehead (widest at the forehead with the apex pointing backward) occurs in 80% to 90% of individuals with piebaldism ( Fig. 11-17 ). Depigmented areas on the forehead often include the whole or inner portions of the eyebrows and eyelashes and extend to the root of the nose. Hypopigmented or depigmented areas have also been noted commonly on the chin, anterior neck, anterior portion of the trunk and abdomen, and on the anterior and posterior aspects of the mid-arm to the wrist and the mid-thigh to mid-calf. Typical of the lesions of piebaldism are islands of normal and increased pigmentation within the hypomelanotic areas and sometimes hyperpigmented borders ( Fig. 11-18 ). Intertriginous freckling and multiple café-au-lait (CAL) macules may occasionally be noted in patients and do not reflect the concurrence of piebaldism and neurofibromatosis (NF) 1 or Legius syndrome.
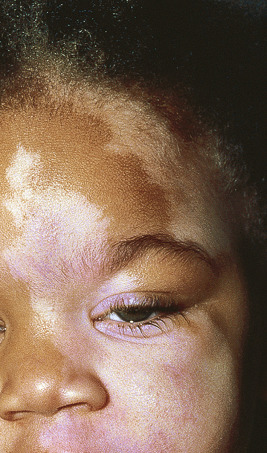
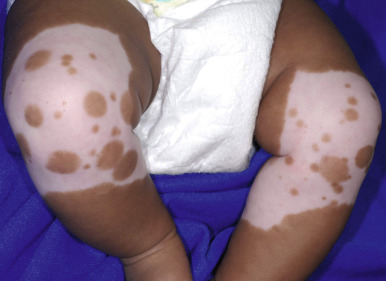
The depigmentation of piebaldism can be differentiated from that of vitiligo by the usual presence at birth, lack of convex borders, and predilection for ventral surfaces in contrast to the predilection on exposed areas, body orifices, areas of trauma, and intertriginous regions in vitiligo. The typical facial characteristics of type I WS are not seen in patients with piebaldism, although sensorineural deafness has rarely been described in piebaldism. Biallelic homozygous mutations in c-KIT have been described from affected consanguineous parents; affected neonates show generalized depigmentation of the skin and hair, blue irides, and profound sensorineural deafness. Incomplete penetrance has been described (e.g., a parent without evidence of piebaldism who has children with piebaldism and shares the c-KIT mutations).
Treatment consists of cosmetic masking of areas of leukoderma and vigorous sun protection. In the rare patients who show increased pigmentation after UV exposure, phototherapy may be considered. Reepithelialization by grafting from suction blisters and autologous cultured or noncultured epidermis, with or without laser, has provided permanent repigmentation.
Waardenburg Syndrome
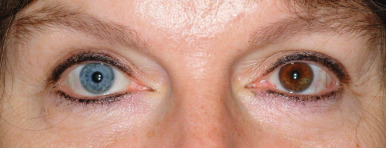
Waardenburg syndrome (WS) is a heterogenous group of autosomal dominant disorders characterized by heterochromia irides, a white forelock, cutaneous depigmentation, and in many patients, congenital sensorineural deafness. WS reportedly accounts for 2% to 5% of cases of congenital deafness. Four major subtypes of WS have been described ( Tables 11-3 and 11-4 ). Individuals with type I WS, the most common form, have characteristic facial features including a broad nasal root and lateral displacement of the medial canthi and lacrimal puncta of the lower eyelids (dystopia canthorum) ( Fig. 11-19 ). For clinical diagnosis, an individual must have two major criteria or one major plus two minor criteria to be considered affected (see Table 11-3 ). Dystopia canthorum can be confirmed by a W index of greater than 1.95 (see Table 11-3 ). Congenital, usually nonprogressive, sensorineural hearing loss occurs in 47% to 58% of affected individuals, whereas the white forelock and cutaneous depigmentation occur in approximately 45% and 30%, respectively. The white forelock may be present at birth, may appear later (typically during teenage years), or may become pigmented with time. The heterochromic irides and/or hypoplastic (often brilliant) blue eyes ( Fig. 11-20 ) are less common than the hair or skin depigmentation. Type I WS results from loss-of-function mutations in PAX3, a gene critical for both melanocyte migration and facial embryogenesis. Spina bifida has been described in several affected families, leading to the firm recommendation for folate supplementation during pregnancy.
| Two Major Criteria or One Major Plus Two Minor Criteria Allows for the Diagnosis of WS Type I | |
|---|---|
| Major Criteria | Minor Criteria |
|
|
* W index: The measurements necessary to calculate the W index (in mm) are as follows: the inner canthal distance (a), the interpupillary distance (b), and the outer canthal distance (c).
| Disorder | Inheritance | Gene | Other Comments |
|---|---|---|---|
| WS1 | AD | PAX3 | Most common form; dystopia canthorum |
| WS2A | AD | MITF | No facial dysmorphism; high risk of hearing loss; iris heterochromia |
| WS2B | AD | ? | No facial dysmorphism; high risk of hearing loss; iris heterochromia |
| WS2C | AD | ? | No facial dysmorphism; high risk of hearing loss; iris heterochromia |
| WS2D | AD | SLUG | No facial dysmorphism; high risk of hearing loss; iris heterochromia |
| WS2E | AD | SOX10 | No facial dysmorphism; high risk of hearing loss; iris heterochromia |
| WS3 | AD | PAX3 | Associated limb abnormalities |
| WS4A | AD/AR | EDNRB | Aganglionic megacolon |
| WS4B | AD/AR | EDN3 | Aganglionic megacolon |
| WS4C | AD | SOX10 | Aganglionic megacolon |
| PCWH | AD | SOX10 | Severe hypotonicity with central nervous system and peripheral nerve abnormalities |
Type II WS is a heterogeneous group of disorders that commonly shows the iris pigmentary changes (almost all patients, particularly the blue irides) and deafness (80%) of WS type I but not the facial characteristics. Mutations in the microphthalmia-associated transcription factor ( MITF ) gene have been described in 15% to 21% of patients with type II WS. Tietze syndrome, also linked to MITF mutations on one allele, was originally characterized by generalized, albino-like pigmentary deficiency and hearing loss but no iris heterochromia; subsequent studies have shown clinical variability within families with heterozygous MITF mutations, ranging from a generalized decrease in pigmentation to patchy loss to no cutaneous manifestations in association with profound hearing loss and blue irides. Patients with WS2 may have strabismus. Freckling in sun-exposed areas without depigmentation is common, especially among patients of Asian descent. Other patients with type II WS have mutations in SOX10 (≈15%, encoding sex-determining region Y [ SRY ]-box10) or SNAI2/SLUG (as in piebaldism), transcription factors critical for the migration and development of neural-crest cells. Type III WS is an extreme presentation of type I WS with musculoskeletal abnormalities and rarely associated neural-tube defects. Some but not all patients with type III WS have homozygous mutations in PAX3 . Type IV WS includes the pigmentary defects and sensorineural deafness in association with absence of enteric ganglia in the distal part of the intestine (Hirschsprung disease); presentation with chronic constipation beginning in the neonatal period is not unusual. Facies are normal. Mutations have been described in three genes: EDN3, EDNRB, and SOX10, encoding endothelin 3, endothelin B receptor, and Sox 10, respectively. Patients with Sox 10 mutations may also have severe hypotonicity with CNS and peripheral-nerve abnormalities because of the important role of Sox 10 in glial cell development. All of the forms of WS show marked variability of clinical characteristics, even within families and in monozygotic twins, and subtle features may be seen, especially in WS1. The white forelock may be a feature of several other genetic and acquired disorders, but most commonly piebaldism. Iris heterochromia has also been described in HPS.
Tuberous Sclerosis Complex
Tuberous sclerosis complex (TSC) is an autosomal dominant disorder with variable expressivity that occurs in as many as 1 in 6000 to 1 in 10,000 persons. Up to 70% of patients are thought to have new mutations. The disorder results from mutation in one of two different genes, TSC1 (encoding hamartin; approximately 20% of patients) and TSC2 (encoding tuberin; approximately 60% of patients); gene mutations have not been discovered in 10% to 25% of affected individuals. Tuberin and hamartin form a complex that suppresses cell growth through regulation of several signaling pathways, most importantly the mammalian target-of-rapamycin (mTOR) pathway signaling through switching Rheb from an active (GTP-bound) to inactive (guanosine diphosphate [GDP]-bound) state. In general, patients with mutations in TSC1 have milder disease. The disorder is characterized by the development of hamartomas of the skin, brain, eye, heart, kidneys, lungs, and bone ( Box 11-1 ). A variety of cutaneous features, including hypopigmented macules, angiofibromas, fibrous tumors, and periungual and gingival fibromas, may be seen.
Pathogenic mutation in TSC1 or TSC2 or two major features or 1 major feature + 2 minor features = definite tuberous sclerosis
One major feature + 1 minor feature = probable tuberous sclerosis
One major feature or two or more minor features = possible tuberous sclerosis
Major Features
Hypopigmented macules ≥5mm across (≥3)
Angiofibromas (>3) or fibrous cephalic plaque
Shagreen patch (connective tissue nevus)
Ungual fibromas (≥2)
Cortical dysplasias
Subependymal nodules
Subependymal giant-cell astrocytoma
Cardiac rhabdomyoma
Multiple retinal hamartomas
Angiomyolipomas (≥2)
Lymphangioleiomyomatosis
Minor features
“Confetti” skin lesions
Intraoral fibromas (>2)
Dental enamel pits (>3)
Multiple renal cysts
Nonrenal hamartomas
Retinal achromic patch
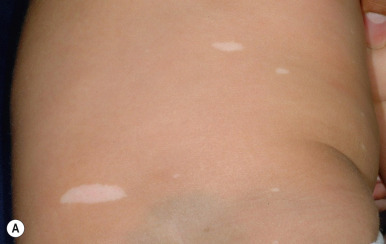
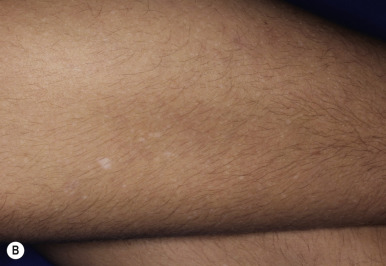
Three or more hypopigmented macules (white spots) of TSC are seen in 97% of patients at birth or shortly thereafter, although the appearance of additional lesions as late as 6 years of age has been described. Once present, the hypopigmented macules tend to be persistent and stable in shape and relative size but may become less apparent during adulthood. Wood lamp examination in a completely darkened room may be useful in accentuating the macules in fair-skinned children. The white spots most commonly occur on the trunk, but hypopigmented tufts of scalp or eyelash hair meet the criterion for a hypopigmented macule. They range in size from a millimeter to several centimeters and number from a few to more than 75; 18% to 20% of individuals with TSC have 1 or 2 hypopigmented macules. The hypopigmented macules ( Fig. 11-21 ) are often round (“thumbprint”), confetti-like hypopigmented macules (particularly over the pretibial areas), and oval or linear, but a lance-ovate shape (“ash leaf spots”) is commonly described and is unusual in other disorders of localized decreased skin pigmentation such as nevus depigmentosus and vitiligo. Lesions of vitiligo tend to be depigmented and show a bright white coloration with Wood lamp examination. The hypopigmented white spots of TSC are most difficult to distinguish from nevus depigmentosus (also called nevus achromicus; Fig. 11-22 ), which occurs in 1.6% to 4.7% of children, suggesting that the majority of young children with a white spot do not have TSC. Nevus depigmentosus may be present at birth or appear during early infancy as normal pigmentation increases and then persist. Despite its name, most nevus depigmentosus lesions are hypopigmented, not depigmented. Most individuals will have a solitary lesion of nevus depigmentosus, but multiple lesions and segmental forms of nevus depigmentosus have been described. Nevus depigmentosus and the hypomelanotic macules of TSC must also be distinguished from nevus anemicus, a developmental anomaly characterized by a circumscribed round or oval patch of pale or mottled skin (see Chapter 12 , Fig. 12-60 ).
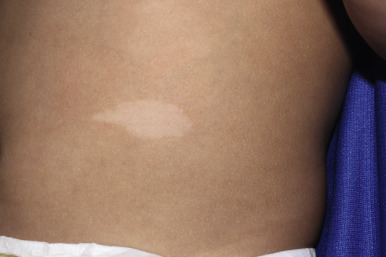
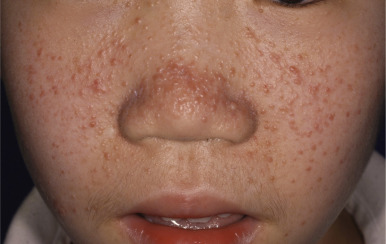
Cutaneous angiofibromas (adenoma sebaceum), which are hamartomas composed of fibrous and vascular tissue, appear in 75% of cases. They typically develop between 2 and 6 years of age and continue to increase in number thereafter but have been described at birth or as late as the mid-20s. These lesions characteristically are 1- to 4-mm, pink to red, dome-shaped papules with a smooth surface. They occur on the nasolabial folds, cheeks, and chin and sometimes more extensively on the face ( Fig. 11-23 ). The upper lip is relatively spared, except immediately below the nose. The angiofibromas in affected adolescents may be masked by or misdiagnosed as acne. Their distribution is usually symmetrical but may be asymmetrical, especially in patients with a mosaic form of TSC.
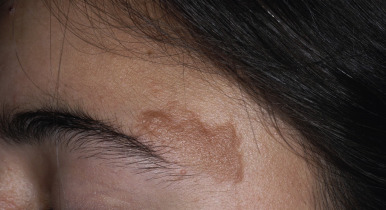
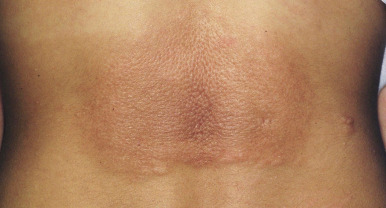
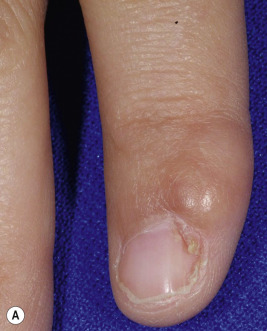
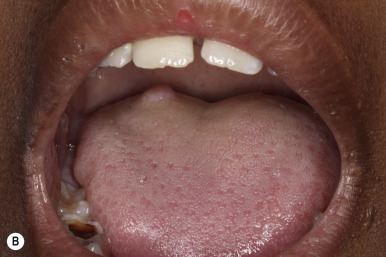
Large fibrotic plaques or nodules may occur on the forehead (fibrous cephalic plaque; Fig. 11-24 ), cheeks, or scalp in 25% of patients and are often present at birth. In 14% to 20% of patients, collagenomas also develop on the trunk, especially in the lumbosacral area, and most commonly during later childhood (shagreen patches or peau de chagrin lesions) ( Fig. 11-25 ). They may be solitary or multiple and vary from smaller than 1 cm to palm-sized. Collagenomas are usually slightly raised with focal depression at follicular openings, leading to comparison with pigskin, elephant skin, orange peel, or gooseflesh. Fibromas under or around the nails of the fingers and especially the toes (ungual fibromas, sometimes called Koenen tumor; Fig. 11-26, A ) and on the gums and other intraoral sites (intraoral fibromas) are also considered pathognomonic ( Fig. 11-26, B ). Seen in up to 80% of patients, these fibromas do not tend to appear until after puberty but may be the only sign of TSC.
Confetti-like hypopigmented macules have been noted in 6% of patients with multiple endocrine neoplasia (MEN) type 1 and multiple angiofibromas in 43% to 88% of adolescents and adults with MEN1. Facial angiofibromas in patients with MEN1 tend to be fewer in number than in patients with TSC, may involve the upper lip or vermilion border, and fail to cluster in the nasolabial folds. Collagenomas have been observed in 72% of patients with MEN1. Patients with MEN1 are at high risk for the development of parathyroid, pituitary, pancreatic, and duodenal tumors. Multiple facial angiofibromas with onset during adulthood have also been noted in patients with Birt–Hogg–Dube syndrome, but facial fibrofolliculomas or trichodiscomas are more typical. Multiple biopsy-proven angiofibromas without other signs and with negative molecular testing have been described. Having one to a few facial fibrous papules is common in normal adults and may begin during adolescence; they resemble angiofibromas and have recently been shown to have activated mTOR signaling as well.
The systemic lesions of TSC may produce severe symptoms and possibly death. Seizures, seen in 80% to 90% of patients with TSC, may begin as infantile spasms in which sudden repetitive myoclonic contractions of most of the body musculature are combined with flexion, extension, opisthotonos, and tremors. By 2 or 3 years of age, focal or generalized seizures and mental retardation may become evident. Extensive CNS involvement leads to hypsarrhythmia (salaam seizures) with electroencephalographic findings of multifocal high-voltage spikes and slow chaotic waves. Later in life the seizure pattern may change to a petit mal variety, and in less severe cases generalized or focal motor seizures may develop.
Retardation may be mild or severe and appears in 62% of affected individuals. The severity of mental retardation correlates well with the age of seizure onset. In approximately 90% of patients with TSC, the brain shows areas of cortical dysplasia (including cortical “tubers” and white-matter migrational abnormalities). These areas of cortical dysplasia can calcify and be visible on skull radiographs as curvilinear opacities. Periventricular or subependymal nodules may be seen by computed tomography (CT) or magnetic resonance imaging (MRI) scanning before calcification occurs. Subependymal nodules are not malignant but may enlarge to cause obstructive hydrocephalus.
By the end of the first decade of life, 80% of patients show renal involvement. Renal hamartomas (angiomyolipomas) occur in about 70% of patients, and larger ones may lead to hemorrhage; of note, angiomyolipomas may be found in other organs as well, and having at least two angiomyolipoma without organ specificity is a major diagnostic feature. With advancing age, 20% to 30% of patients develop multiple bilateral renal cysts resembling those of polycystic kidney disease. These cysts can occur in individuals with TSC1 or TSC2 mutations, but a subset of individuals with aggressive renal cysts have deletions involving both TSC2 and the contiguous polycystic kidney disease ( PKD1 ) genes. Abdominal ultrasound or scans are able to detect renal hamartomas or cysts in asymptomatic patients. Cardiac rhabdomyomas are most commonly present in the ventricles prenatally or in infancy and tend to regress spontaneously. Although usually asymptomatic, rhabdomyomas may be associated with congestive heart failure, murmurs, cyanosis, arrhythmias, and sudden death, particularly during the first year of life. Two-dimensional echocardiography is a noninvasive technique that allows detection of asymptomatic cardiac rhabdomyomas.
The eyes may have characteristic retinal lesions (gliomas) referred to as phakomas. These retinal hamartomas have been described in 30% to 50% of patients. Funduscopy may show one of two types of lesions: multiple, raised, mulberry-like lesions on or adjacent to the optic nerve head; or flat, disk-like lesions in the periphery of the retina. Pulmonary lymphangioleiomyomatosis occurs overall in 2.3% of individuals with TSC, particularly in women between the ages of 20 and 40 years. Affected individuals experience shortness of breath, hemoptysis, or pneumothorax and show diffuse interstitial infiltrates with cystic changes by CT examination. About 85% of patients with TSC have osseous manifestations with the bones, particularly those of the hands and feet, demonstrating cysts and periosteal thickenings. At least three dental pits (seen as punctate, round or oval, 1- to 2-mm randomly arranged enamel defects), particularly in the permanent teeth, are another marker of TSC and may be visualized better with use of dental disclosing solution.
The diagnosis of TSC may be difficult, because many affected individuals have subtle manifestations. However, the appearance of characteristic skin lesions in children with seizures, retardation, or both should establish a diagnosis of TSC (see Box 11-1 ). Diagnosis depends on the cutaneous manifestations, family history (with careful clinical and sometimes imaging examination of family members), MRI of the brain, renal ultrasound, cardiac echocardiography in infants and young children, and in some cases ophthalmologic examination and chest radiography for honeycombing of the lungs.
The prognosis of TSC depends on the severity of the disorder and the presence of neurologic involvement. The leading causes of premature death, status epilepticus and bronchopneumonia, are related to the associated neurologic issues. Seizures can be controlled by anticonvulsant therapy in many patients, and prevention of seizures early in life has been shown to lower the risk of developmental delay and retardation. With routine MRI evaluations and the availability of microscopic surgery for neoplastic brain lesions, patients are surviving longer and have a better quality of life. Neurosurgical intervention may be required in patients with signs of increased intracranial pressure such as visual disturbances, papilledema, vomiting, or headaches. Sun protection is important for patients, not just at the sites of the hypopigmented macules, but also to prevent facial angiofibromas, which show second-hit mutations in TSC genes with a UV-signature mutation that is not seen in TSC germline mutations, in 50% of patients.
The facial angiofibromas may be a cosmetic problem that responds to cryosurgical, surgical, or laser therapy. However, a meta-analysis of 16 reports noted improvement in 94% of 84 patients with use of a variety of formulations of compounded topical rapamycin (ointment, gel, solution, cream; there is no commercially available formulation) with concentrations from 0.003% to 1%). Recent-onset fibrous angiofibromas respond best, and topical rapamycin has also been used for maintenance after laser or surgical therapy. Local irritation is unusual but has been described with the use of rapamycin solution. Topical rapamycin has also led to improvement in early ungual fibromas. Mammalian TOR inhibitors such as rapamycin and everolimus cause regression of astrocytomas, renal angiomyolipomas, and pulmonary lymphangioleiomyomas, as well as facial angiofibromas. The Tuberous Sclerosis Alliance (at www.tsalliance.org ) and the Tuberous Sclerosis Association (of the United Kingdom, at www.tuberous-sclerosis.org ) are among the groups that offer support for patients with TSC.
Chemically Induced Depigmentation
A number of chemical agents are known to cause depigmentation after topical exposure. Among these compounds are the rubber antioxidant monobenzyl ether of hydroquinone, hydroquinone photographic developer, sulfhydryl compounds, azo dyes, diphenylcyclopropenone, phenolic germicidal agents (paratertiary butylphenol and amylphenol), hydroxyanisole, and 4-tertiary butyl catechol (an additive to polyethylene film). The biochemical mechanism by which phenolic chemicals induce such hypopigmentation appears to be the competitive inhibition of tyrosinase or the release of toxic metabolites that produce injury to the melanocytes. Depigmentation has also occurred after topical exposure to paraphenylenediamine, injections of triamcinolone, and in the periorbital area after injection of botulinum A toxin. Oral ingestion of chloroquine, arsenic, STI571 (imatinib), and dasatinib have also led to depigmentation. A progressive generalized decrease in pigmentation has been reported after drug reaction to sulfonamide.
Idiopathic Guttate Hypomelanosis
Idiopathic guttate hypomelanosis is a common disorder of adults, and its incidence increases with increasing age. It occasionally occurs in children and is more common in female individuals, although the latter may represent reporting bias. More striking in individuals with darker pigmentation, the lesions of idiopathic guttate hypomelanosis are characteristically 0.5- to 6-mm sharply defined, porcelain white macules. They are asymptomatic and once present do not tend to change. The macules most commonly occur on the extensor forearms and on the shins. The diagnosis is usually made clinically, and no treatment is effective. The underlying cause is unknown, although sun exposure is thought to be a trigger.
Postinflammatory Hypopigmentation
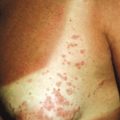
Postinflammatory hypopigmentation (or leukoderma) may be associated with a wide variety of inflammatory dermatoses or infections. This relative pigmentary deficiency may be noted after involution of certain inflammatory skin disorders, particularly burns, bullous disorders, infections, eczematous or psoriatic lesions, and pityriasis rosea (see Fig. 3-17 ). In the inflammatory dermatoses the intensity of the inflammatory reaction may bear little relationship to the development of postinflammatory leukoderma. Postinflammatory hypopigmentation is generally self-limiting, clearing after months to years. It often becomes cosmetically obvious in individuals with darker skin, particularly during summer months, because the preferential darkening with UV light exposure of surrounding skin accentuates the hypopigmentation.
Although the pathophysiology of postinflammatory hypopigmentation is unclear, it is postulated that the hypopigmentation is caused when keratinocytes injured by the inflammatory process are temporarily unable to accept melanosomes from the melanocyte dendrites. No therapy is effective, but the condition tends to improve with time.
Pityriasis alba (see Chapter 3 ) is a common cutaneous disorder characterized by asymptomatic, sometimes scaly hypopigmented patches on the face, neck, upper trunk, arms, shoulders, and at times the lower aspect of the trunk and extremities of children and young adults (see Fig. 3-33 ). Seen predominantly in children 3 to 16 years of age, individual lesions vary from 1 to several centimeters in diameter and have sharply delineated margins and a fine, branny scale. Although the cause is unknown, this disorder appears to represent a nonspecific dermatitis. Postinflammatory hypopigmentation is commonly seen in children with atopic dermatitis, psoriasis (see Fig. 4-15, A ), pityriasis lichenoides, and contact dermatitis; in the latter the pattern of the hypopigmentation may provide the clue to the contactant.
Tinea versicolor (pityriasis versicolor; see Chapter 17 ) is a common condition often found on the upper part of the trunk and neck of young adults. Caused by overgrowth of a yeast that normally inhabits skin, Pityrosporum orbiculare ( Malassezia furfur ), the condition is characterized by small, either hypopigmented or occasionally hyperpigmented macules, particularly on the trunk and upper arms. The round, individual lesions often coalesce. Facial involvement is more common in affected children than in older individuals. The hypopigmentation results from the production of azelaic acid, which inhibits tyrosinase; the hyperpigmentation is postinflammatory (see Fig. 17-33 , Fig. 17-34 ).
Sarcoidosis (see Chapter 25 ) is a granulomatous disorder of unknown origin with widespread manifestations involving the skin and many of the internal organs. In addition to the characteristic yellowish brown, flesh-colored, pink, red, and reddish brown to black or blue lesions, subcutaneous nodules, and infiltrated plaques, the spectrum of sarcoidal skin lesions includes hypomelanotic macules and papules. Measuring up to 1.5 cm in diameter, these hypopigmented lesions reveal sarcoid-type granulomas on cutaneous biopsy.
Leprosy (Hansen disease; see Chapter 14 ), a chronic infection in which the acid-fast bacillus Mycobacterium leprae has a special predilection for the skin and nervous system, can be divided into several types depending on the patient’s cellular immune response to M. leprae . Tuberculoid leprosy shows characteristic well-defined anesthetic hypopigmented lesions and thickened and palpable peripheral nerves. Lepromatous leprosy, in contrast, more commonly shows nodules or diffuse infiltrates, especially on the eyebrows and ears, resulting in a leonine facies. A granulomatous infiltrate on microscopic examination of cutaneous lesions and particularly in the lepromatous lesions, demonstration of M. leprae on cutaneous smear or biopsy specimen generally confirm the diagnosis.
Pinta (see Chapter 14 ) is a treponemal infection caused by Treponema carateum . Seen almost exclusively among the dark-skinned population of Cuba and Central and South America, the disorder is commonly found in children of parents afflicted with this disorder. The cutaneous manifestations may be divided into primary, secondary, and tertiary stages. The late dyschromic stage takes several more years to develop. These lesions have an insidious onset and usually appear during adolescence or young adulthood. They consist of slate-blue hyperpigmented lesions that after a period of years become widespread and are replaced by depigmented macules resembling those seen in patients with vitiligo. Located chiefly on the face, waist, and areas close to bony prominences (elbows, knees, ankles, wrists, and the dorsal aspect of the hands), these depigmented lesions of pinta can be differentiated from those of other depigmented disorders by the presence of pigmented lesions, histologic examination of lesional specimens, identification of antibodies directed against T. carateum by serologic testing, and darkfield examination.
Disorders of Both Hypopigmentation and Hyperpigmentation
Dyschromatoses
Two major forms of dyschromatoses have been described: dyschromatosis symmetrica hereditaria (DSH; reticulate acropigmentation of Dohi) and dyschromatosis universalis hereditaria (DUH), both of which are seen most commonly in Japanese and Chinese individuals (in about 2 per 100,000 individuals). These disorders show only pigmentary manifestations and affected individuals are almost always otherwise healthy. The reticulate hyperpigmentation of these disorders tends to appear more spotty than the more net-like reticulated hyperpigmentation of disorders like dyskeratosis congenita (see Chapter 7 ), Rothmund–Thomson (see Chapter 19 ) and Kindler (see Chapter 13 ) syndromes. The differential diagnosis of these dyschromatoses includes other disorders with more of a macular pigmentation (such as xeroderma pigmentosum (see Chapter 19 ), Kitamura reticulate acropigmentation, and dyschromic amyloidosis ( Table 11-5 ). Keratin disorders with hyperpigmentation also tend to have a more net-like reticulated pigmentation (e.g., EB simplex with mottled pigmentation [see Chapter 13 ], Dowling–Degos disease, Naegeli–Franceschetti–Jadassohn syndrome and dermatopathia pigmentosa reticularis).
| Disorder | Inheritance | Gene | Onset | Features | Other Comments |
|---|---|---|---|---|---|
| Familial forms of amyloidosis | Unknown | Early childhood | Generalized hyperpigmentation with tiny generalized hypopigmented macules | AD form: focal subepidermal amyloid deposition in biopsies | |
| Amyloidosis cutis dyschromica | AD | Carriers of X-linked form: along lines of Blaschko, resembling incontinentia pigmenti | |||
| X-linked reticulate hyperpigmentation | XL | XL form: failure to thrive, developmental delay, seizures, hemiplegia, colitis, gastroesophageal reflux, inguinal hernia, urethral stricture, dental anomalies, hypohidrosis, photophobia, corneal clouding, skeletal changes | XL form: often no amyloid in biopsies of children | ||
| Dowling–Degos disease | AD | Keratin 5; POFUT1 ; POGLUT1 | Early adolescence | Reticulate pigmentation of flexures, neck, and sometimes generalized; sometimes pitted perioral scars and comedo-like follicular plugs | No acantholysis More generalized reticulated pigmentation in EBS with mottled pigmentation ( KRT5 mutations; see Ch. 13 ) |
| Familial progressive hyperpigmentation and hypopigmentation | AD | KITLG | Birth or early infancy; increase in number with time | Face, neck, trunk, limbs with reticulate hyperpigmentation as well as diffuse background hyperpigmentation; café-au-lait macules and freckling; sometimes larger hypopigmented macules | Called familial progressive hyperpigmentation if no hypopigmentation; distinguish from NF1 and Legius syndrome |
| Dyschromatosis symmetrica hereditaria | AD | ADAR1 | Infancy to early childhood | Hyperpigmented and hypopigmented small macules on dorsum of hands and feet | |
| Dyschromatosis universalis hereditaria | AD | ABCB6 | First months of life (starts on trunk) | Generalized pigmented macules; may involve palms and soles, oral mucosa, and nails (dystrophy with pterygium) | |
| Galli–Galli disease | AD | Keratin 5 | Early adolescence | Reticulate pigmentation of flexures | Moderate to severe suprabasal acantholysis in biopsies |
| Kitamura disease | AD | ADAM10 | First to second decade | Acral reticular pigmentation with subtle atrophy, palmar pits, and rete ridge breaks | |
| Dyskeratosis congenita (see Ch. 7 ) | Esp. XL | Esp. DKC1 | Late childhood | Net-like pigmentation, esp. in sun-exposed areas, often poikilodermatous; associated with mucosal leukokeratosis, nail dystrophy, risk of bone marrow failure, mucosal squamous cell carcinomas | Nine known genes can be mutated; can be AD or AR |
| Naegeli–Franceschetti–Jadassohn syndrome | AD | KRT14 | First 2 years | Reticulate hyperpigmentation, primarily of abdomen, perioral, and periocular areas; palmoplantar keratoderma, absence of dermatoglyphics, onychodystrophy, hypohidrosis, dental anomalies Pigmentation fades during adolescence | |
| Dermatopathia pigmentosa reticularis | AD | KRT14 | Pigmentation primarily truncal distribution; nonscarring alopecia; palmoplantar keratoderma, absence of dermatoglyphics, onychodystrophy, hypohidrosis |
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


