Abstract
Disorders of hyperpigmentation usually result from an increase in melanin production, which is occasionally associated with an increased density of active melanocytes. Skin discoloration may also be caused by dermal deposition of exogenous substances such as drugs or heavy metals. Clinically, disorders of hyperpigmentation can be classified into diffuse, circumscribed, linear, and reticulated subsets. This chapter reviews disorders within each of these categories, including both common and rare conditions. A discussion of dyschromatoses, disorders characterized by both hypo- and hyperpigmentation, follows.
Keywords
diffuse hyperpigmentation, flagellate pigmentation, reticulated hyperpigmentation, linear hyperpigmentation, postinflammatory hyperpigmentation, dyschromatosis, melasma, erythema dyschromicum perstans, pigmentary demarcation lines, pigmentary mosaicism, flagellate pigmentation, prurigo pigmentosa, dyskeratosis congenital, Naegeli–Franceschetti–Jadassohn syndrome, dermatopathia pigmentosa reticularis, X-linked reticulate pigmentary disorder, Dowling–Degos disease, reticulate acropigmentation of Kitamura, dyschromatosis symmetrica hereditaria, dyschromatosis universalis hereditaria
Skin color depends upon the amount and distribution of melanin and other pigments such as hemoglobin, which can influence light absorption, reflection, and scattering. Disorders of hyperpigmentation usually result from an increase in melanin production and, on occasion, from an increase in the density of active melanocytes. Discoloration of the skin may also be caused by deposition of exogenous substances such as drugs, drug complexes (e.g. with melanin or iron), or heavy metals within the dermis.
To aid in the clinical approach, disorders of hyperpigmentation can be divided into diffuse, circumscribed, linear, and reticulated subsets ( Fig. 67.1 ). A discussion of each of these categories is followed by an overview of dyschromatoses, disorders characterized by both hypo- and hyperpigmentation. Benign melanocytic neoplasms are covered in Chapter 112 .
Diffuse and Circumscribed Hyperpigmentation
Introduction
Diffuse and circumscribed hyperpigmentation are discussed together because both postinflammatory hyperpigmentation (PIH) and reactions to systemic drugs, two major causes of hyperpigmentation, can assume either of these patterns. PIH most often presents as circumscribed lesions, and the size, shape, and distribution pattern provide clues to the etiology. Melasma is another common cause of circumscribed hypermelanosis. Diffuse hyperpigmentation can also be a manifestation of metabolic diseases, sclerodermoid disorders, nutritional deficiencies, and occasionally HIV infection.
Postinflammatory Hyperpigmentation
▪ Postinflammatory hypermelanosis
- ▪
Extremely common, especially in individuals with more darkly pigmented skin
- ▪
Develops after inflammation or injury to the skin, but preceding inflammation may be transient or subclinical
- ▪
The increased melanin may be primarily within the dermis (e.g. following lichen planus) or in the epidermis (e.g. following acne or atopic dermatitis)
- ▪
Epidermal hyperpigmentation fades more readily than dermal hyperpigmentation
Introduction
PIH represents an acquired excess of melanin pigment following cutaneous inflammation or injury. It can occur anywhere on the skin surface, including the mucous membranes and the nail unit. PIH is extremely common and can have significant cosmetic and psychosocial consequences.
Epidemiology
PIH can occur at any age and there is no gender preference. Individuals with darkly pigmented skin tend to have a greater frequency, severity, and duration of PIH than those with lighter complexions.
Pathogenesis
In the epidermal form of PIH, there is increased melanin production and/or transfer to keratinocytes. Inflammatory mediators (e.g. prostaglandins E 2 and D 2 ) that enhance pigment production may play a role in this process. In dermal hyperpigmentation, melanin enters (“falls into”) the dermis via a damaged basement membrane, where it is phagocytosed by and subsequently resides within dermal macrophages (referred to as melanophages). Macrophages may also migrate into the epidermis, phagocytose melanosomes, and then return to the dermis. Melanin within dermal melanophages tends to persist for long periods of time (e.g. years).
Clinical Features
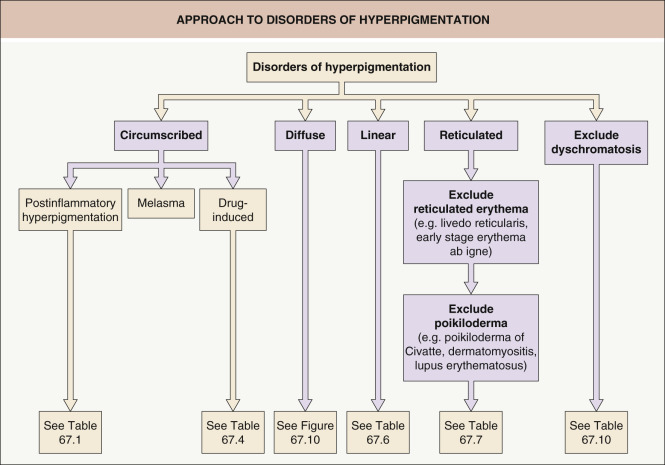
Asymptomatic hyperpigmented macules and patches range in color either from tan to dark brown (epidermal melanin) or from gray–blue to gray–brown (dermal melanin). Primary lesions of the underlying inflammatory disorder may or may not be evident admixed with the hyperpigmentation or elsewhere. When primary lesions are absent, the size, shape, and distribution pattern of the hyperpigmented lesions may provide clues to the underlying etiology ( Table 67.1 ). PIH can be exacerbated by continued inflammation, trauma, exposure to ultraviolet (UV) irradiation, or treatment-related irritation.
| DISORDERS ASSOCIATED WITH POSTINFLAMMATORY HYPERPIGMENTATION | |
|---|---|
| Inflammatory disease | Clinical clues |
| Common | |
| Acne vulgaris | Head/neck region, upper trunk; <1 cm; perifollicular |
| Atopic dermatitis | Atopic diathesis; face and extensor extremities in infants, then later flexural involvement; excoriations; atopic pleats; xerosis; lichenification; transverse nasal crease (“allergic salute”) |
| Impetigo | Favors face; most common in children |
| Insect bites | Favor exposed areas; usually <1 cm; lower extremities common with flea bites; clustered and sometimes linear patterns (“breakfast–lunch–dinner”; see Fig. 67.2B ) |
| Lichen simplex chronicus | Common locations: posterior neck, ankle, scrotum |
| Transient neonatal pustular melanosis | Black newborns; pustules precede pigmentation (see Fig. 67.2C ) |
| Less common | |
| Irritant and allergic contact and photocontact dermatitis | Sites determined by etiologic agent and form of exposure; phytophotodermatitis associated with linear hyperpigmentation in sun-exposed areas |
| Pityriasis rosea | Favors trunk and proximal extremities; lesions follow skin cleavage lines; oval-shaped |
| Psoriasis | Scalp/nail involvement; knees/elbows most common sites (see Fig. 67.2D ) |
| Seborrheic dermatitis | Favors face (especially forehead, upper eyelids, nasolabial folds, mental fold), skin folds, and presternal area; scalp/ear involvement; may also have hypopigmentation |
| Polymorphous light eruption | Extensor upper extremities, mid upper chest, face; seasonal (e.g. spring or early summer) |
| Discoid lupus erythematosus | Face and conchal bowls, with follicular plugging in latter site; oral lesions; in scarred lesions, central hypopigmentation with rim of hyperpigmentation |
| Lichen planus | Wrists, shins, presacral; nail/oral involvement |
| Erythema dyschromicum perstans (EDP; ashy dermatosis) * | Neck, proximal upper extremities, trunk; round or oval in shape with gray–brown to blue–gray color; long axis can follow skin cleavage lines (similar to pityriasis rosea); less commonly observed in fair-skinned individuals |
| Idiopathic eruptive macular pigmentation * | Trunk, proximal extremities, neck, face; round or oval, discrete brown macules and small patches; sometimes barely elevated plaques with a subtle velvety texture; histologically, basal epidermal hyperpigmentation ± papillomatosis, with no vacuolar alteration and relatively few dermal melanophages compared to EDP; affects primarily children and adolescents, lasting months to years |
| Fixed drug eruption | Circular; favors perioral, acral and genital sites; recurrence at same site(s) with repeated exposure (see Fig. 67.2E ) |
| Morbilliform drug eruption | Widespread; usually discrete lesions, history of drug exposure |
| Viral exanthem | Widespread; usually discrete lesions; history of associated symptoms |
| Morphea | Trunk or extremities; large-sized except in guttate variant; may be linear; associated induration and later dermal atrophy |
| Atrophoderma of Pasini and Pierini | Trunk; large-sized; depressed with “cliff sign” at periphery no induration |
| Neurotic (psychogenic) excoriation, acne excoriée | Favors face, scalp, extensor surface of arms, upper back (reachable areas); linear or angular shapes; multiple stages of evolution, from erosions/ulcerations to scars |
| Flagellate erythema | Associated with bleomycin use, shiitake mushroom ingestion, dermatomyositis, and Still disease |
* Typically no preceding inflammatory phase evident clinically or histologically.
Disorders that commonly lead to epidermal PIH include acne, insect bites, pyodermas, atopic dermatitis, psoriasis, and pityriasis rosea ( Fig. 67.2A–D ). In contrast, dermal PIH is associated with dermatoses characterized by degeneration of the basal layer of the epidermis and inflammation at the dermal–epidermal junction, such as lichen planus, lichenoid drug reactions, lupus erythematosus, and fixed drug eruptions ( Fig. 67.2E ). Upon treatment of the underlying disorder, epidermal PIH generally resolves over time, although fading may require months or years in darkly pigmented individuals. Dermal melanosis tends to persist longer and is sometimes permanent.
Pathology
Epidermal PIH is characterized by increased pigment in keratinocytes and dermal PIH by melanophages within the dermis.
Differential Diagnosis
The size, shape, and distribution of the hyperpigmented lesions can provide clues to the inflammatory disease or injury that preceded PIH (see Table 67.1 ), and a thorough skin examination may detect active primary lesions. A history of prior inflammatory lesions and medication use (including prescription, over-the-counter, and alternative products) should be obtained. Disorders such as erythema dyschromicum perstans, melasma, pityriasis versicolor, and atrophoderma of Pasini and Pierini should be considered in patients without evidence of preceding inflammation by history or on examination. Occasionally, a biopsy may assist in establishing the diagnosis.
Treatment
Provided that the underlying dermatosis is successfully treated, PIH eventually improves in most patients, especially those with epidermal hypermelanosis. Sun protection, including daily application of a broad-spectrum sunscreen, can help to prevent accentuation of the pigmentation by UV exposure. Topical hydroquinone (2–4%) may lead to lightening over 3–6 months if the increase in pigmentation is limited to the epidermis. As with melasma (see below), use of a topical retinoid and corticosteroid together with hydroquinone may be more effective than monotherapy. Topical azelaic acid, α-hydroxy acid, L-ascorbic acid, and kojic acid as well as arbutin, licorice extracts, mequinol, niacinamide, N -acetyl glucosamine, and soy are additional potential therapeutic options . However, it is important to avoid irritant contact dermatitis from any topical agent. Chemical peels (e.g. with salicylic or glycolic acid) and laser therapy may be of benefit but can also result in hypopigmentation, especially in patients with darkly pigmented skin. Q-switched ruby, alexandrite, and Nd:YAG lasers are variably successful in removing dermal pigment (see Ch. 137 ).
Erythema Dyschromicum Perstans
▪ Ashy dermatosis ▪ Dermatosis cenicienta
- ▪
Most common in individuals with skin phototypes III–IV
- ▪
Gray–brown to blue–gray macules and patches in a symmetric distribution
- ▪
Favors the neck, trunk, and proximal extremities
- ▪
A consistently effective treatment is not currently available
Introduction
Ramirez first described erythema dyschromicum perstans (EDP) in 1957, referring to affected individuals as los cenicientos (ashen ones). This is an asymptomatic, slowly progressive eruption that is characterized by circumscribed areas of dermal pigmentation .
Epidemiology
Although EDP occurs worldwide, it is most common in Latin America. There is no gender preference. The disorder usually presents during the second to third decade, but it occasionally develops in younger children or older adults.
Pathogenesis
The etiology of EDP is not known. Although it has been postulated that a cell-mediated immune reaction to an ingestant, contactant, or microorganism underlies the discrete areas of pigmentary incontinence, no causal agent has consistently been identified . In most patients, a trigger is never found. However, there have been reports of EDP developing in association with the ingestion of ammonium nitrate, oral X-ray contrast media, and medications (e.g. benzodiazepines, penicillin); exposure to various pesticides, fungicides or toxins; endocrinopathies such as thyroid disease; and whipworm and HIV infections. The presence of the HLA-DR4 allele may represent a risk factor for EDP in Mexican patients.
Clinical Features
Oval, circular, or irregularly shaped macules and patches that are 0.5 to 3 cm in diameter and slate-gray to blue–brown in color develop gradually in a symmetric pattern and may coalesce ( Fig. 67.3A ). The initial site of involvement is often the trunk, with subsequent spread to the neck, proximal upper extremities, and sometimes the face. The mucous membranes are spared, and the palms, soles, and scalp are rarely affected. Occasionally, early lesions have a thin, raised, erythematous border, which tends to resolve over a few months, and the long axis of lesions may follow skin cleavage lines. Peripheral hypopigmentation may be seen in older lesions.
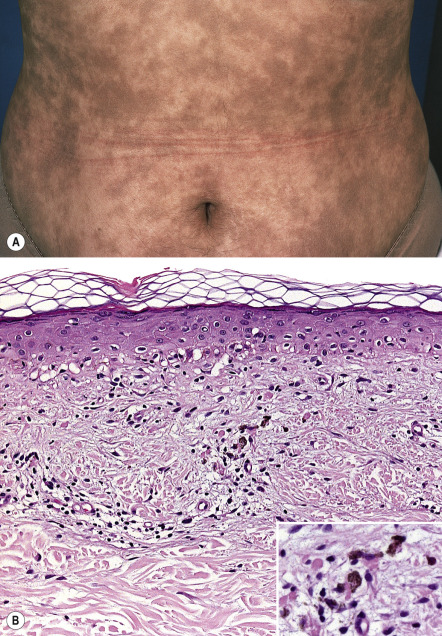
EDP is usually asymptomatic but can be mildly pruritic. The disease progresses slowly and typically does not regress in adults. However, in one study of 33 prepubertal children, the majority of whom were Caucasian, the disease spontaneously resolved in two-thirds of the patients, with no recurrences over the ensuing 2–3 years .
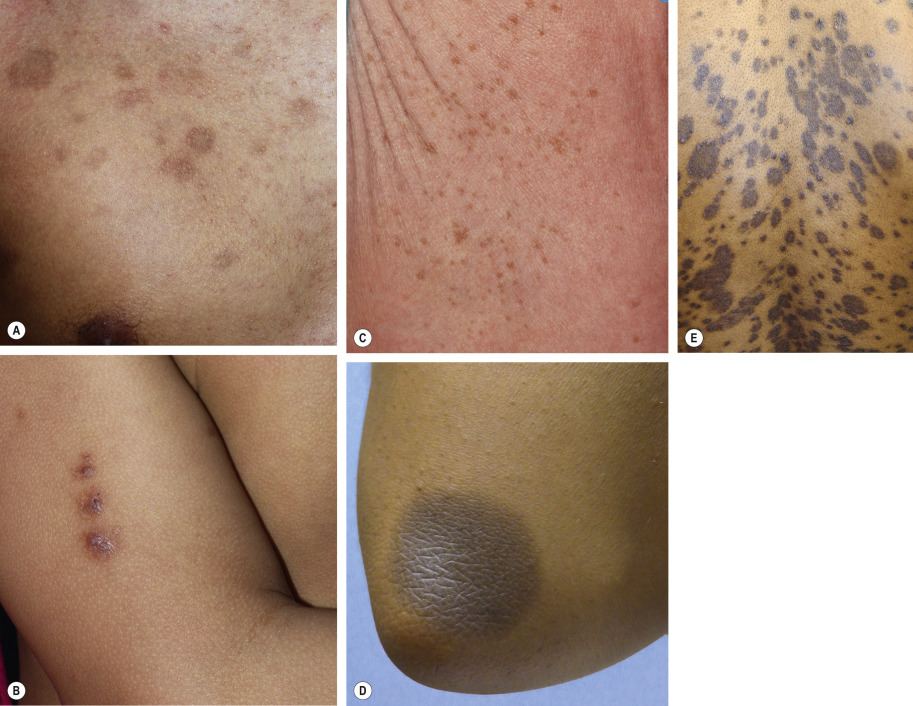
Pathology
In early (“active”) lesions, vacuolar degeneration of the basal cell layer, a perivascular mononuclear cell infiltrate and melanophages in the upper dermis, and increased epidermal melanin are seen ( Fig. 67.3B ). Colloid bodies and dermal hemosiderin may be present. Immunohistochemical findings include expression of cell adhesion and lymphocyte activation molecules (e.g. CD36, intercellular adhesion molecule-1 [ICAM-1], CD69, CD94) that are associated with inflammation and activation of cytotoxic T cells. In later (“inactive”) lesions, the number of melanophages in the superficial dermis is increased but hydropic changes within the basal cell layer are absent and the dermal mononuclear cell infiltrate is minimal or absent.
Differential Diagnosis
Idiopathic eruptive macular hyperpigmentation (IEMH) features brown macules and patches with a similar size and distribution as in EDP but primarily epidermal pigment; recent studies have emphasized a velvety surface and papillomatosis histologically (see Table 67.1 ). The differential diagnosis of EDP also includes a lichenoid drug eruption (see Ch. 11 ), lichen planus (especially lichen planus pigmentosus), generalized fixed drug eruption, mastocytosis, and (less often) infectious diseases such as leprosy or pinta.
Treatment
There is no consistently effective therapy for EDP. Topical corticosteroids and hydroquinone are generally of no benefit. Topical tacrolimus and oral corticosteroids, antibiotics, antimalarials, isoniazid, and griseofulvin, as well as UV light therapy, have produced variable results. Successful treatment with dapsone and clofazimine has been reported in small series.
Lichen Planus Pigmentosus
- ▪
Variant of lichen planus most commonly described in adults with skin phototypes III–V
- ▪
Gray–brown to dark brown macules and patches either in a photodistribution or in an inverse (intertriginous) pattern
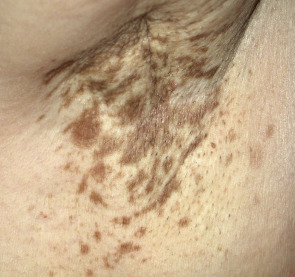
The etiology of LPP is unknown. The photodistribution in some patients suggests that UV can play a pathogenic role, and topical application of mustard oil (which contains allyl isothiocyanate, a potential photosensitizer) and amla oil have been proposed as possible inciting agents . The differential diagnosis includes the actinic and inverse variants of lichen planus (see Ch. 11 ), a lichenoid drug eruption, EDP, melasma and the entities listed in Table 67.2 when facial, PIH, and cutaneous T-cell lymphoma. In one uncontrolled study, lightening of the pigmentation occurred in 54% (7/13) of patients treated with topical tacrolimus for 12–16 weeks .
| DIFFERENTIAL DIAGNOSIS OF MELASMA | |
|---|---|
| Disorder | Key differences from melasma/comments |
| Drug-induced hyperpigmentation or discoloration |
|
| Postinflammatory hyperpigmentation, e.g. due to cutaneous lupus erythematosus, photosensitivity reactions, contact dermatitis (e.g. irritant contact dermatitis to hydroquinone or tretinoin), or other forms of dermatitis |
|
| Pigmented contact dermatitis (Riehl melanosis) |
|
| Acquired bilateral nevus of Ota-like macules (Hori nevus) |
|
| Actinic lichen planus |
|
| Lichen planus pigmentosus |
|
| Erythema dyschromicum perstans |
|
| Exogenous ochronosis |
|
| Cutaneous mercury deposits |
|
| Erythromelanosis follicularis faciei et colli |
|
| Poikiloderma of Civatte |
|
| Acanthosis nigricans |
|
Melasma
▪ Chloasma ▪ Mask of pregnancy
- ▪
At least 90% of patients are women
- ▪
Increased prevalence in individuals who are Hispanic, or of Asian or African descent
- ▪
Most common location is the face, followed by the forearms
- ▪
Symmetric patches of hyperpigmentation with irregular borders due to increased melanin within the epidermis and/or dermis
Introduction and Epidemiology
Melasma is a common acquired disorder characterized by symmetric, hyperpigmented patches with an irregular outline, occurring most commonly on the face. It is most prevalent among young to middle-aged women who are Hispanic or of Asian, African, or Middle Eastern descent. Exacerbating factors include sun exposure, pregnancy, and use of oral contraceptives.
Pathogenesis
Although the exact pathogenesis of melasma is unknown, it is hypothesized that following exposure to UV irradiation or another inducer, hyperfunctional melanocytes within involved skin produce increased amounts of melanin . The key role of UV irradiation is supported by fading of lesions during winter months and a distribution pattern characterized by involvement of sun-exposed regions and sparing of relatively sun-protected sites such as the philtrum. In addition to oral contraceptive use and hyperestrogenic states, other medications (e.g. phenytoin, phototoxic drugs) and disorders (e.g. autoimmune thyroid disease) have the potential to aggravate melasma. Increased expression of KIT and stem cell factor within the lesional epidermis and dermis, respectively, may play a role in the hyperpigmentation of melasma .
Clinical Features
Light to dark brown or brown–gray patches with irregular borders appear primarily on the face ( Fig. 67.5 ). The areas of hypermelanosis are distributed symmetrically in three classic patterns: (1) centrofacial (most common), involving the forehead, cheeks, nose, upper lip (sparing the philtrum and nasolabial folds), and chin; (2) malar , affecting the cheeks and nose; and (3) mandibular , along the jawline. Less common sites include the extensor aspect of the forearms and mid upper chest. Lesions often first appear or are accentuated following exposure to UV irradiation or during pregnancy. In lightly pigmented individuals, this “mask of pregnancy” frequently diminishes or disappears after parturition, but it tends to persist in women with more darkly pigmented skin.
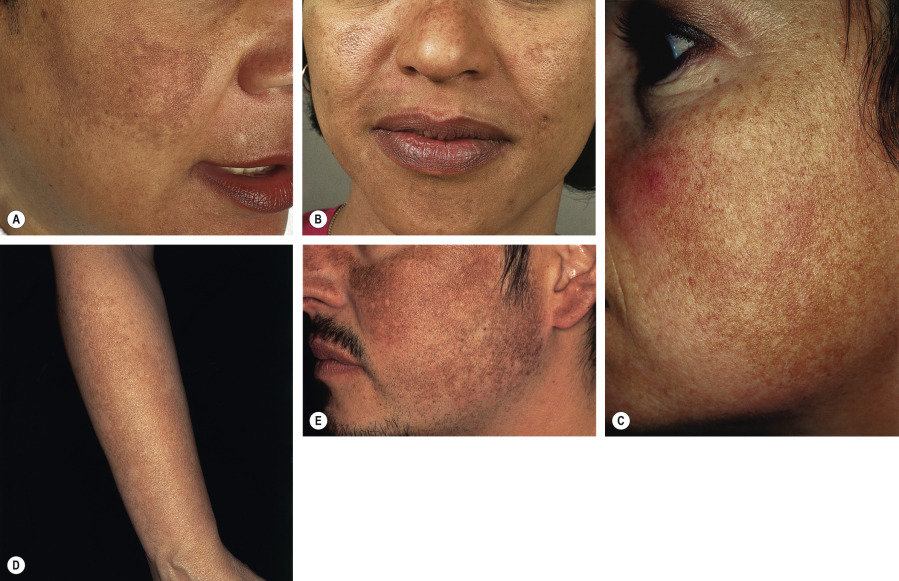
Melasma has classically been subdivided into four types based upon the primary location of the pigment: epidermal, dermal, mixed, or indeterminate (e.g. in patients with very dark skin pigmentation). In theory, lesions with increased epidermal melanin are accentuated and those with increased dermal melanin become less obvious (i.e. blend with uninvolved skin) with Wood’s lamp examination. However, clinicopathologic studies utilizing adjacent uninvolved skin as controls have shown that Wood’s lamp examination does not correlate with histologic findings . In addition, no completely dermal form of melasma was observed histologically when bilateral nevus of Ota-like macules, a form of dermal melanocytosis sometimes misdiagnosed as melasma (see Table 67.2 ), were excluded. Considering that epidermal pigmentation is more likely to respond to topical therapies, further studies are needed to determine the clinical utility and prognostic significance of Wood’s lamp examination.
Pathology
Compared to uninvolved adjacent skin, increased melanin deposition is observed in all layers of the epidermis, particularly the basal layer. An increased number of melanophages may also be seen in the upper dermis. Epidermal melanocytes are normal to slightly increased in number, and they are enlarged with prominent dendrites .
Ultrastructurally, lesional melanocytes contain an increased number of melanosomes. In addition, the mitochondria, Golgi apparatus, and rough endoplasmic reticulum are increased in number or amount. These findings support the theory of hyperfunctional melanocytes, presumably stimulated by UV irradiation or hormones (see Pathogenesis above).
Differential Diagnosis
The differential diagnosis of melasma is reviewed in Table 67.2 . These disorders are distinguished from melasma based upon historical aspects (e.g. drug ingestion, previous inflammation), color, distribution pattern, histologic features, and, if present, primary inflammatory lesions.
Treatment
The treatment of melasma is summarized in Table 67.3 . Diligent sun protection and patient motivation are necessary for any melasma treatment regimen to be successful. For epidermal melasma, 2 months of therapy are typically required to initiate lightening and 6 months of treatment are often needed to achieve satisfactory results.
| TREATMENT OPTIONS FOR MELASMA |
| Recommendations for all patients |
|
| Active treatment * , ** |
|
| Long-term maintenance |
|
* Results from topical treatments may take up to 6 months to appreciate; depending on the patient, HQ or a combination HQ + retinoid + corticosteroid are typically used daily for 2–4 months and then decreased in frequency to 1–2 times per week; prolonged daily use can result in side effects such as perioral dermatitis and atrophy (corticosteroid) or exogenous ochronosis (HQ; see Ch. 129 ).
** While topical HQ can cause allergic contact dermatitis, all topical agents may lead to irritant contact dermatitis, which can worsen the dyspigmentation; if this is a concern, can test on a small, non-facial site prior to widespread facial application.
§ Typically a class 5–7 topical corticosteroid is used (see Ch. 125 ).
¶ Potential risk of post-procedural dyspigmentation; a small test site should be performed prior to widespread facial laser or light therapy.
Additional Forms of Circumscribed Hyperpigmentation
Segmental Pigmentation Disorder
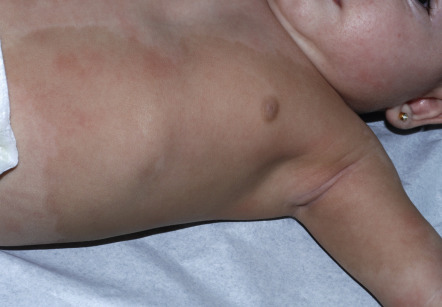
This term was coined by Metzker et al. in 1983 and re-introduced by Hogeling and Frieden in 2010 . It is a type of pigmentary mosaicism, i.e. a postzygotic genetic alteration leading to a regional change in pigment production potential, that is characterized by a block-like pattern of hyper- or (less often) hypopigmentation ( Fig. 67.6 , see Ch. 62 ). The hyperpigmented patches are evident at birth or become apparent during infancy. They favor the trunk, with midline demarcation more often evident ventrally than dorsally and less distinct lateral borders. There are usually no associated extracutaneous anomalies.
Familial Progressive Hyperpigmentation
Familial progressive hyperpigmentation is an autosomal dominant disorder characterized by hyperpigmented patches in a widespread distribution including the palms, soles, lips, and conjunctiva. The lesions begin to develop during infancy and increase in size, number, and confluence with age. Heterozygous gain-of-function mutations in the KIT ligand gene ( KITLG ) can cause this condition as well as familial progressive hyper- and hypopigmentation , which has the additional finding of hypopigmented macules and patches.
Primary (Localized) Cutaneous Amyloidosis
Macular and lichenoid forms of primary (localized) cutaneous amyloidosis are associated with hyperpigmentation (see Ch. 47 ). The most common locations are the upper back (macular amyloidosis) or the extensor surface of the lower extremities (lichen amyloidosis), and there is a characteristic rippled pattern with parallel bands or ridges of hyperpigmentation. Areas of involvement are often pruritic, and rubbing plays a key role in the production of lesions. Histologically, melanophages as well as amyloid deposits that stain positively with anti-keratin antibodies are seen within the upper dermis.
Mastocytosis
Cutaneous mastocytosis is a spectrum of disorders characterized by the accumulation of mast cells within the skin and sometimes in other organs (see Ch. 118 ). Urticaria pigmentosa, the maculopapular form of mastocytosis, classically features hyperpigmented lesions, with patients developing a few to several hundred brown to red–brown macules and papules. However, solitary mastocytomas can also have a light brown color. Lesions of cutaneous mastocytosis typically urticate after they are stroked (Darier’s sign), and an increased number of mast cells is seen in biopsy specimens. The cause of the hyperpigmentation in mastocytosis may involve stimulation of melanocytes by mast cell-derived mediators such as KIT ligand (stem cell factor) or α-melanocyte-stimulating hormone . Clonal somatic activating mutations in the gene that encodes the KIT receptor often underlie mastocytosis in both children and adults. Whereas the prognosis for childhood-onset mastocytosis is good, with spontaneous resolution by adolescence in many patients, adult-onset disease is frequently persistent (see Ch. 118 ).
Tinea (Pityriasis) Versicolor
Tinea versicolor is a superficial skin infection caused by the yeast Malassezia globosa and other Malassezia spp. (see Ch. 77 ). Hypo- or hyperpigmented macules or very thin papules and plaques covered with fine scale are usually found on the upper trunk and proximal upper extremities but may also appear in other sites such as the neck, face, and groin. There is often coalescence of lesions centrally, especially on the trunk. In light and electron microscopy studies, a similar number of melanocytes was noted in uninvolved skin as in hypo- and hyperpigmented lesions; the latter had a thickened stratum corneum that contained more organisms. The clinical differential diagnosis for the hyperpigmented form of pityriasis versicolor may include PIH and confluent and reticulated papillomatosis, but a KOH preparation of the scale showing both hyphal and yeast forms of Malassezia distinguishes tinea versicolor from these entities.
Atrophoderma of Pasini and Pierini
Atrophoderma of Pasini and Pierini is usually seen in young adults and has been postulated to represent an atrophic form of morphea (see Ch. 99 ). Most often, multiple oval hyperpigmented patches measuring 4–10 cm in diameter appear on the posterior trunk. There is a subtle depression of the entire lesion, but no induration or secondary changes. The depression can be appreciated by palpation of the edge of the lesion, with a characteristic “cliff sign” at the peripheral margin.
Pigmented Lesions
Lentigines
Lentigines are reviewed in Chapters 109 and 112 .
Café-au-Lait Macules
Café-au-lait macules (CALMs) are hyperpigmented macules or patches two to three shades darker than uninvolved skin (see Ch. 112 ). They are found in at least 10% of the population. Although evident at birth in ~3% of all neonates and up to 30% of black neonates, CALMs may not become apparent until infancy or early childhood in lightly pigmented individuals. The presence of multiple CALMs raises the possibility of neurofibromatosis type 1 or another genetic syndrome (see Table 61.4 ); Fig. 61.10 outlines an approach to a young child with multiple CALMs. A CALM is usually easily distinguished from a fully developed congenital melanocytic nevus, but in infants there may be overlap in the clinical appearance of these two entities. The differential diagnosis also includes the earliest stage of a nevus spilus (before the “speckles” appear), Becker melanosis (nevus) or smooth muscle hamartoma, and a mastocytoma; in addition, segmental CALMs and “pigmentary mosaicism” have significant overlap. Although laser treatment of CALMs is possible (see Ch. 137 ), responses vary and recurrence of pigmentation is common.
Drug-Induced Hyperpigmentation or Discoloration
- ▪
Localized or generalized hypermelanosis or discoloration may be seen
- ▪
Several medications and drug classes are known to induce hyperpigmentation or discoloration, especially minocycline, antimalarials, chemotherapeutic agents, and zidovudine
- ▪
Longitudinal, transverse, or diffuse melanonychia may also be present
| DRUGS AND CHEMICALS ASSOCIATED WITH HYPERPIGMENTATION OR DISCOLORATION | ||
|---|---|---|
| Drug or chemical | Clinical features | Histopathology/comment |
| Chemotherapeutic agents | ||
| BCNU (carmustine) |
|
|
| Bleomycin (intravenous or intralesional) |
|
|
| Busulfan |
|
|
| Cyclophosphamide |
|
|
| Dactinomycin |
|
|
| Daunorubicin |
| |
| Doxorubicin |
|
|
| 5-Fluorouracil |
|
|
| Hydroxyurea |
|
|
| Mechlorethamine (nitrogen mustard) |
|
|
| Methotrexate |
|
|
| Antimalarials | ||
| Chloroquine, hydroxychloroquine, quinacrine |
|
|
| Heavy metals | ||
| Arsenic |
|
|
| Bismuth |
|
|
| Gold (chrysiasis) |
|
|
| Iron |
|
|
| Lead |
|
|
| Mercury |
|
|
| Silver (argyria) |
|
|
| Hormones | ||
| Oral contraceptives |
|
|
| ACTH/MSH (ACTH is rarely used; [NIe4-D-Phe7]-α-MSH/afamelanotide orphan drug for EPP) |
|
|
| Miscellaneous compounds | ||
| Amiodarone |
|
|
| Azidothymidine (zidovudine, AZT) |
|
|
| Clofazimine |
|
|
| Diltiazem (rarely amlodipine) |
|
|
| Dioxins |
|
|
| Ezogabine |
|
|
| Hydroquinone |
|
|
| Imatinib (also dasatinib) |
|
|
| Minocycline |
|
|
| Psoralens |
|
|
| Psychotropic drugs (chlorpromazine, thioridazine, imipramine, desipramine, amitriptyline) |
|
|
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


