Abstract
Dermatomyositis is an autoimmune disease that most often affects both the muscles and the skin (classic dermatomyositis), but also may occur in a skin-predominant fashion (clinically amyopathic dermatomyositis). All adult patients presenting with cutaneous lesions of dermatomyositis warrant investigation for concomitant muscle disease, systemic involvement, and/or malignancy, whereas children have no increased risk of cancer. Despite pathognomonic and characteristic cutaneous findings, the diagnosis of dermatomyositis is often delayed, particularly in the absence of muscle disease. The cutaneous findings of dermatomyositis and the corresponding histopathologic features may be mistaken for lupus erythematosus. One helpful clinical clue, if present, is marked pruritus which can impact the patient’s quality of life. Treatment recommendations for cutaneous dermatomyositis, based primarily upon case series or retrospective reviews, include photoprotection, topical corticosteroids, and oral antimalarials. For more severe disease, an immunosuppressive or immunomodulatory agent such as methotrexate, mycophenolate mofetil or intravenous immunoglobulin, can be employed.
Keywords
dermatomyositis, myositis, heliotrope, Gottron papules, photodistributed poikiloderma, inflammatory myopathy, amyopathic dermatomyositis, interstitial lung disease, paraneoplastic, calcinosis cutis, holster sign, Gottron sign, shawl sign
- ▪
Autoimmune connective tissue disease of uncertain etiology demonstrating a bimodal age distribution with both juvenile and adult forms
- ▪
Clinical and laboratory signs of proximal extensor inflammatory myopathy
- ▪
Distinctive, photodistributed, pink–violet poikiloderma favoring the scalp, periocular region, and extensor surfaces, in addition to distinctive nail-fold changes and pink–violet papules overlying the knuckles
- ▪
Histopathologic evidence of interface dermatitis plus mucin deposition in skin biopsy specimens and lymphocytic myositis in biopsy specimens from affected muscle
- ▪
When myositis is present, therapy includes systemic corticosteroids and oftentimes steroid-sparing immunosuppressive drugs, with the prognosis being very good, except for those individuals with refractory myositis, systemic involvement, or an associated advanced malignancy
- ▪
Management of cutaneous dermatomyositis via skin-directed therapies can be challenging and is directed by disease severity
Introduction
Along with polymyositis and inclusion body myositis, dermatomyositis is classified as one of the idiopathic inflammatory myopathies. Dermatomyositis is a disease of presumed autoimmune pathogenesis that presents with a symmetric, proximal, extensor inflammatory myopathy and a characteristic cutaneous eruption. Previously, polymyositis was thought to represent the same disease process but limited to the muscles; however, there is now evidence that the pathogenetic mechanisms of polymyositis and dermatomyositis differ significantly. In polymyositis, clonally expanded, autoreactive CD8 + T cells invade myocytes expressing MHC class I antigens and cause necrosis via the perforin pathway. In contrast, there are data to support the theory that autoantigens activate a humoral immune process in dermatomyositis, in which complement is deposited in capillaries causing capillary necrosis and ischemia . Because an appreciation of these pathogenetic differences occurred relatively recently, previous studies often pooled cases of dermatomyositis and polymyositis, making interpretation of their data challenging.
Dermatomyositis is characterized by a bimodal age distribution, with both adult and juvenile forms, and up to one-quarter of patients in the adult group having an associated malignancy. Patients with juvenile dermatomyositis do not have an increased risk of malignancy, but do have an increased incidence of calcinosis cutis and associated small vessel vasculitis. There is a subset of patients in whom the cutaneous manifestations of dermatomyositis exist without objective evidence of muscle inflammation, referred to as amyopathic dermatomyositis, and formerly known as dermatomyositis sine myositis. For dermatologists, establishing the diagnosis of dermatomyositis is important because it is a serious, treatable, multisystem disease that has a different prognosis and treatment approach from lupus erythematosus (LE). The malignancy association in adults provides the dermatologist with an opportunity to assist medical colleagues in detecting the tumor at an early stage because the skin disease often precedes the onset of symptoms related to the malignancy.
History
Polymyositis and dermatomyositis have been recognized as clinical entities for more than 100 years. Although the first adult patients with dermatomyositis and malignancy were described early in the twentieth century, a causal association was not suggested until the 1940s. A major step forward in the understanding of these disorders occurred in 1975, when Bohan and Peter proposed generally accepted clinical criteria for their diagnosis . Over the past 15 years, it has become more widely accepted that a subset of patients has skin-limited disease (i.e. amyopathic dermatomyositis). For this reason, Sontheimer proposed a revised classification of the idiopathic inflammatory myopathies that includes amyopathic forms of dermatomyositis, with muscle involvement no longer required for a definitive diagnosis ( Table 42.1 ) . Based upon recent epidemiologic studies, an estimated 20% of dermatomyositis patients have clinically amyopathic disease .
| REVISED CLASSIFICATION SYSTEM FOR THE IDIOPATHIC INFLAMMATORY DERMATOMYOPATHIES |
|
| Inclusion body myositis |
* Both adult-onset and juvenile-onset amyopathic DM and hypomyopathic DM can be further subcategorized as “provisional” and “confirmed” when patients have biopsy-confirmed hallmark cutaneous manifestations of DM without muscle weakness and with normal muscle enzymes for ≥6 months (provisional) or 24 months (confirmed).
Epidemiology
Dermatomyositis is a relatively rare disease that occurs throughout the world. In adults, dermatomyositis affects women two to three times more often than men. The incidence of dermatomyositis ranges from 2 to 9 per million amongst various populations and seems to be increasing, although this may simply be a result of increased detection, as well as better diagnosis and reporting. Table 42.2 summarizes the findings in four recent studies .
| CLINICAL SUBTYPES OF DERMATOMYOSITIS – DEMOGRAPHICS AND ASSOCIATED FINDINGS | ||||
|---|---|---|---|---|
| Clinical subtype | ||||
| Adult-onset classic dermatomyositis | Adult-onset clinically amyopathic dermatomyositis | Juvenile-onset classic dermatomyositis | Juvenile-onset clinically amyopathic dermatomyositis | |
| No. of patients | 20 | 291 | 120 | 38 |
| Mean age (years) | 51.9 | 50 | 7.7 | 10.5 |
| Malignancy detected | 6/20 (30%) | 41/291 (14%) | 0/120 (0%) | 0/38 (0%) |
| Sex ratio (F : M) | 3 : 1 | 3 : 1 | 2.2 : 1 | 3 : 2 |
Pathogenesis
Dermatomyositis is believed to result from an immune-mediated process triggered by “outside” factors (e.g. malignancy, drugs, infectious agents) in genetically predisposed individuals. Table 42.3 illustrates some of the evidence that supports this theory . Serum antinuclear autoantibodies are often present, as are other myositis-specific autoantibodies, as summarized in Table 42.4 . The antisynthetase antibodies are directed against cytoplasmic antigens; therefore the antinuclear antibody test may be negative. Patients with antisynthetase antibodies often have overlap syndromes. The term antisynthetase syndrome has been coined to refer to patients with these autoantibodies, fever, erosive polyarthritis, “mechanic’s hands”, Raynaud phenomenon, and interstitial lung disease .
| PATHOGENESIS OF DERMATOMYOSITIS |
| Genetics |
|
| Cellular immunity/apoptosis |
|
| Humoral immunity |
|
| Infectious precipitants |
|
| Drug and vaccine precipitants |
|
| Malignancy association (adults) |
|
| SERUM AUTOANTIBODIES IN ADULT AND JUVENILE DERMATOMYOSITIS | ||||
|---|---|---|---|---|
| Autoantibodies | Target antigen function | Clinical phenotype | Autoantibody frequency, % | |
| Adult DM | Juvenile DM | |||
| Anti-aminoacyl-tRNA synthetases (e.g. anti-Jo-1 [histidyl], anti-PL-7 [threonyl]; see Ch. 40 ) | Intracytoplasmic protein synthesis | Antisynthetase syndrome (myositis, mechanic’s hands, erosive polyarthritis, fever, Raynaud phenomenon, high frequency of interstitial lung disease) | up to 20 | 1–3 |
| Anti-SRP | Protein translocation | Acute-onset necrotizing myopathy (severe weakness, high CK); may be refractory to treatment | 5 | <1 |
| Anti-Mi-2 | Helicase – transcription | Adult DM and juvenile DM (hallmark is cutaneous disease, milder muscle disease with good response to treatment) | 15 | <10 |
| Anti-TIF-1γ (p155) | Cell proliferation, apoptosis, innate immunity | Cancer-associated myositis in adult DM; extensive cutaneous disease in adult DM and juvenile DM; palmar hyperkeratotic papules, psoriasiform lesions, red-on-white telangiectatic patches, ovoid palatal patch | 80 (amyo) | ~25 |
| 20–40 (classic) | ||||
| Anti-NXP-2 (p140) | Nuclear transcription, RNA metabolism | Cancer-associated myositis in adult DM; adult and juvenile DM with calcinosis; peripheral edema, myalgia, dysphagia, mild skin disease | 10–20 | ~25 |
| Anti-SAE | Post-translational modification | Adult DM; may present with clinically amyopathic DM | 5 | NA |
| Anti-MDA5 (CADM-140) | Innate immunity | Cutaneous ulcerations – periungual region and overlying Gottron papules and/or Gottron sign, oral ulcerations, prominent alopecia, arthritis, clinically amyopathic DM, interstitial lung disease including a rapidly progressive form | 5–20 | NA |
Clinical Features
Cutaneous Disease
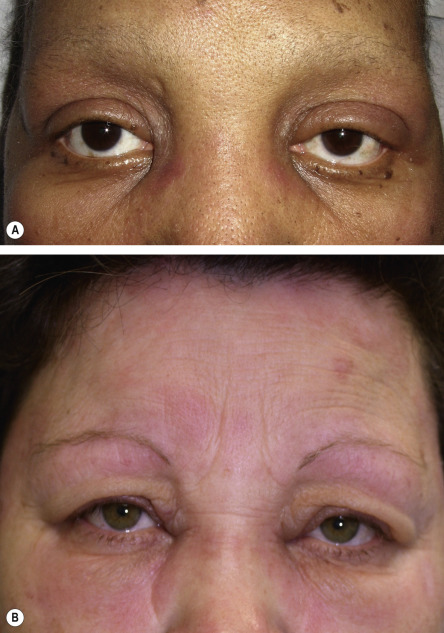
The features that are classically considered pathognomonic for dermatomyositis include the heliotrope sign and Gottron papules ( Fig. 42.1 ). The former is characterized by a pink–violet color, primarily of the eyelids and periorbital skin, and there may be associated edema ( Fig. 42.2 ). The heliotrope sign can be quite subtle, with only mild erythema of the eyelids, and it may wax and wane in intensity. Some patients will have more widespread facial erythema or midfacial erythema that tends to involve the nasolabial folds ( Fig. 42.3 ).
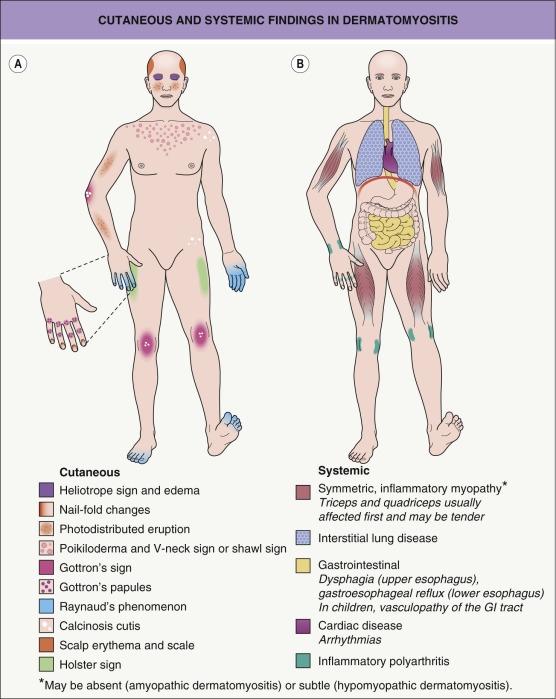
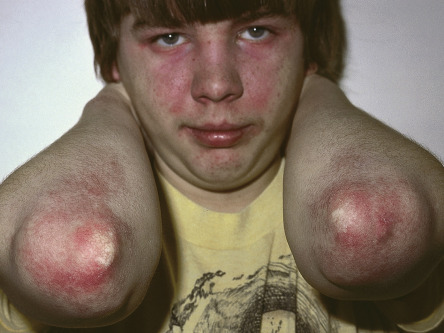
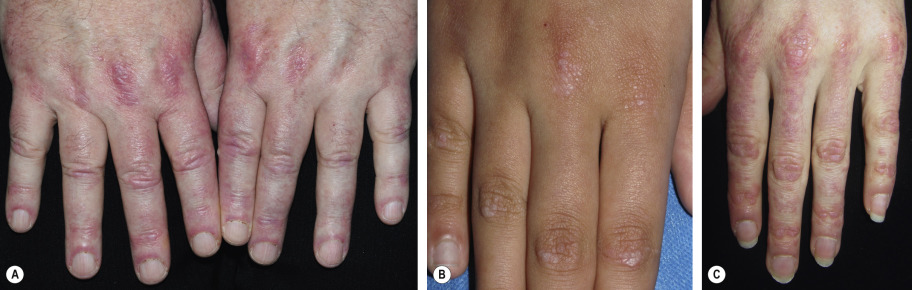
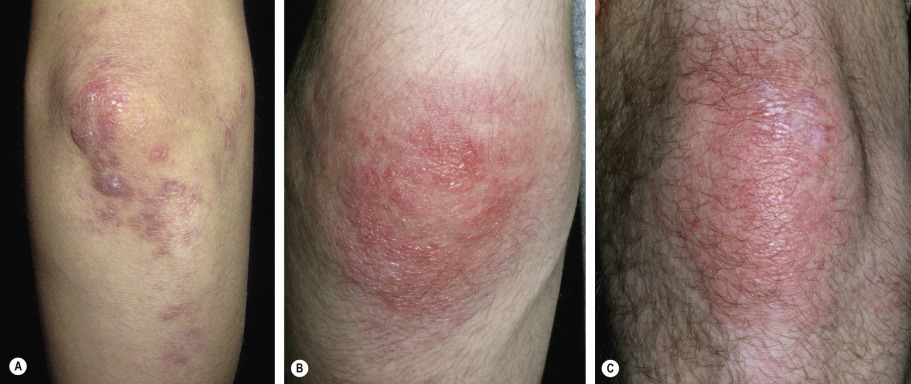
The cutaneous lesions of dermatomyositis are accentuated on extensor surfaces, including the elbows, knees, metacarpophalangeal joints, and both the proximal and distal interphalangeal joints (knuckles; Fig. 42.4A ). When papules on the knuckles develop a secondary lichenoid quality, they are termed Gottron papules ( Fig. 42.4B,C ); involvement of the elbows and/or knees is known as Gottron sign ( Fig. 42.5 ).
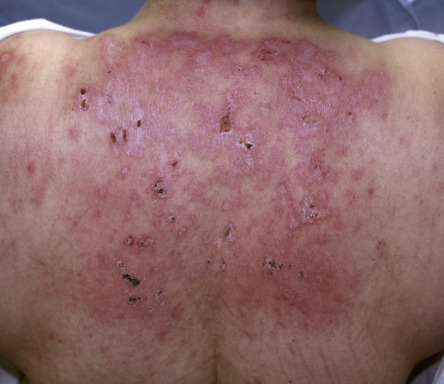
A critical diagnostic feature of the cutaneous eruption of dermatomyositis is poikiloderma. This feature may occur both in patients with dermatomyositis, where it is frequently characterized by a pink–violet color, and in patients with lupus erythematosus, where the poikiloderma is a more red color. However, in patients with skin phototypes I and II, the hue of the poikiloderma of dermatomyositis may be pink–red rather than truly violet. Photodistributed poikiloderma is very characteristic of dermatomyositis, often involving the upper chest ( V-neck sign ) and the upper back ( shawl sign ). Poikiloderma can also affect photoprotected areas, such as the lateral thigh, referred to as the holster sign . If the clinician misses the poikiloderma, the eruption of dermatomyositis may occasionally be misdiagnosed as psoriasis because there may be lesions on the elbows and knees as well-defined plaques with fine silvery scale.
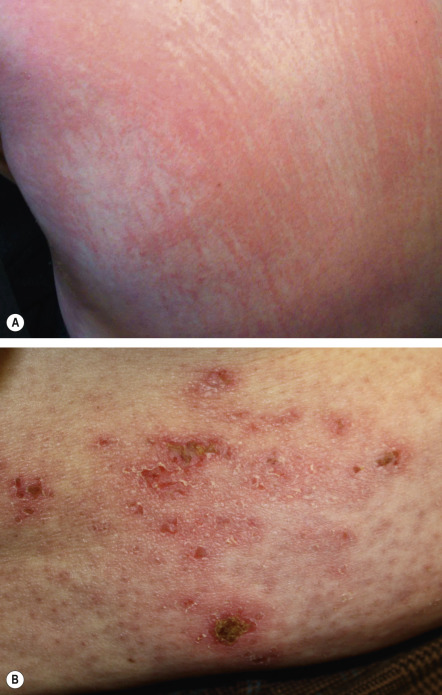
The cutaneous lesions of dermatomyositis are often intensely pruritic ( Fig. 42.6 ), and this can significantly affect patients’ quality of life . Of note, pruritus is a feature that can occasionally help to distinguish dermatomyositis from LE.
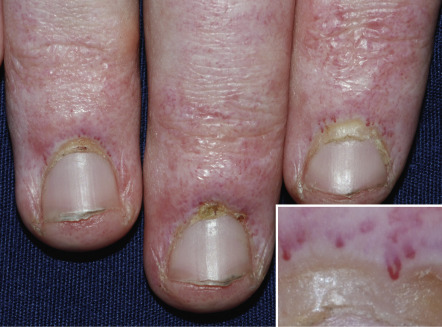
An additional clinical clue to the diagnosis of dermatomyositis is nail-fold changes. Cuticular dystrophy (i.e. “ragged” cuticles and cuticular hypertrophy) is quite characteristic, as are nail-fold telangiectasias in which dilated capillary loops alternate with capillary dropout ( Fig. 42.7 ). If the photodistribution and nail-fold changes are missed, the eruption may be misdiagnosed as other conditions characterized by poikiloderma, such as cutaneous T-cell lymphoma. Often, the dermatologist correctly notices the photodistribution but considers the diagnosis of photodrug eruption or lupus erythematosus rather than dermatomyositis.
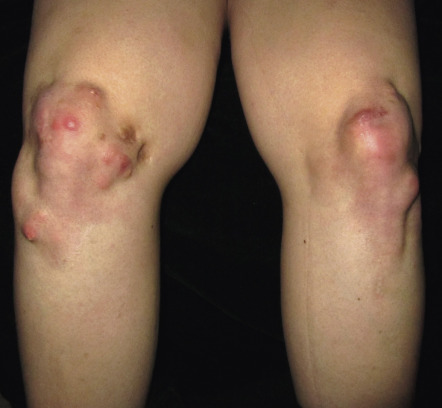
Calcinosis cutis is more prevalent in juvenile dermatomyositis, affecting 25–70% of pediatric patients , and it is usually associated with a delay in diagnosis, a delay in the institution of systemic corticosteroids, and/or advanced, therapy-resistant disease. In addition to the skin, calcium deposits may develop within the deep fascia and intramuscular connective tissue. Calcinosis cutis presents as hard, irregular papules or nodules that occasionally drain a chalky material (see Ch. 50 ). Lesions favor sites of trauma, such as the elbows and knees ( Fig. 42.8 ), but can occur anywhere and may be painful and interfere with function.
Additional cutaneous manifestations range from poikiloderma and scaling of the scalp (often accompanied by non-scarring alopecia) to centripetal flagellate erythema ( Fig. 42.9A ) to erosions and ulcerations ( Fig. 42.9B ; Table 42.5 ) . Wong-type dermatomyositis is a variant seen more commonly in Asians in which the cutaneous findings mimic pityriasis rubra pilaris . Lastly, because of the frequency of overlap syndromes, it is important to look for the skin signs of other autoimmune connective tissue diseases in patients with dermatomyositis.
| CUTANEOUS MANIFESTATIONS OF DERMATOMYOSITIS | |
|---|---|
| Common | Uncommon |
|
|
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


