Abstract
Because of the frequency of skin involvement in systemic diseases, cutaneous manifestations often prove very helpful in establishing the correct diagnosis. The skin signs of systemic disease can be specific, e.g. cutaneous lupus, cutaneous sarcoidosis, in part because of associated histologic features, while others are best described as suggestive, e.g. pyoderma gangrenosum, acquired ichthyosis. Cutaneous findings that raise the possibility of an underlying malignancy, referred to as paraneoplastic, can serve as an early warning sign, appearing prior to the diagnosis of the associated malignancy. In general, paraneoplastic skin diseases improve if the associated malignancy responds to appropriate therapy and their recurrence should prompt a thorough search for recurrence of the cancer. In this chapter, the characteristic dermatologic manifestations of rheumatologic, hematologic/oncologic, gastrointestinal and cardiopulmonary disorders are reviewed, with cross references to more detailed descriptions in chapters throughout the book.
Keywords
cutaneous manifestations of systemic diseases, rheumatologic disease, endocrine disease, metabolic syndrome, hematologic disease, paraneoplastic skin diseases, paraneoplastic dermatoses, skin signs of systemic disease, acanthosis nigricans, gastroenterology and skin disorders, peristomal skin disorders, necrolytic migratory erythema, rheumatoid arthritis
Keywords
cutaneous manifestations of systemic diseases, rheumatologic disease, endocrine disease, metabolic syndrome, hematologic disease, paraneoplastic skin diseases, paraneoplastic dermatoses, skin signs of systemic disease, acanthosis nigricans, gastroenterology and skin disorders, peristomal skin disorders, necrolytic migratory erythema, rheumatoid arthritis
- ▪
Because of the frequency of skin involvement in systemic diseases, cutaneous manifestations often prove helpful in establishing the correct diagnosis
- ▪
Skin signs of systemic disease can be specific, e.g. cutaneous lupus, cutaneous sarcoidosis, in part because of associated histologic features, while others are best described as suggestive, e.g. pyoderma gangrenosum, acquired ichthyosis
- ▪
Cutaneous disease can serve as the initial manifestation of a systemic disorder, including an internal malignancy
Skin disease is often associated with internal manifestations , e.g. psoriatic arthritis, and there are many cutaneous disorders that are traditionally associated with systemic disease. For example, rheumatologic disorders frequently have skin manifestations, as do infectious diseases and endocrinopathies. When one examines many of the disorders that are discussed elsewhere in this text, it becomes clear that isolated dermatologic disease is relatively uncommon. A detailed review of all dermatologic signs of internal disease is beyond the scope of this chapter. Rather, representative disorders are discussed individually, while the remainder are reviewed via tables that contain information relevant to dermatologists. Disorders are grouped on the basis of affected organ systems.
Cutaneous Rheumatology
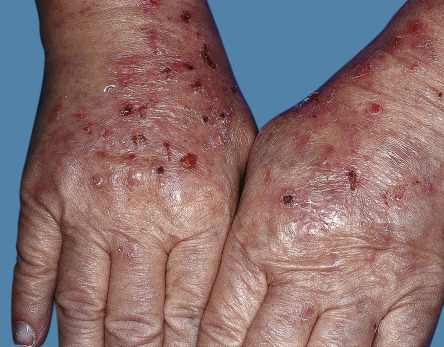
The spectrum of disorders traditionally considered to be cutaneous rheumatology includes: lupus erythematosus ( Ch. 41 ); dermatomyositis ( Ch. 42 ); systemic sclerosis ( Ch. 43 ); vasculitides ( Ch. 24 ); and miscellaneous disorders. Miscellaneous disorders usually include: rheumatoid arthritis ( Ch. 45 ), reactive arthritis (formerly Reiter disease) ( Ch. 8 ), psoriatic arthritis ( Ch. 8 ), Behçet disease ( Ch. 26 ), pyoderma gangrenosum ( Ch. 26 ), Sweet syndrome ( Ch. 26 ), bowel-associated dermatosis–arthritis syndrome ( Ch. 26 ), Kawasaki disease ( Ch. 81 ), relapsing polychondritis ( Ch. 45 ), and several of the autoinflammatory disorders (see Table 45.7 ).
Cutaneous conditions reported in patients with rheumatoid arthritis are summarized in Table 53.1 ( Figs 53.1–53.5 ) . Dermatologists should be particularly careful in this group of patients to couple laboratory evaluation with a comprehensive cutaneous examination, including all mucosal surfaces, nails, nail folds, and hair. Clinical criteria are published for the diagnosis of many of these disorders; dermatologists should be familiar with these criteria and incorporate them into a focused, thorough history, which includes relevant negatives. It is very helpful to consider the clinicopathologic basis and pathogenesis of cutaneous lesions. For example, patients with systemic lupus erythematosus can have different types of photodistributed lesions with an interface histopathology, e.g. discoid, subacute, poikilodermatous, and they have different implications with regard to systemic disease. Such lesions are also treated differently than those caused by vessel-based pathology, e.g. small or larger vessel vasculitis.
| CUTANEOUS MANIFESTATIONS OF RHEUMATOID ARTHRITIS | |
|---|---|
| Condition | Features |
| Palisading granulomas | |
| Rheumatoid nodules |
|
| Interstitial granulomatous dermatitis (IGD) and palisaded neutrophilic & granulomatous dermatitis (PNGD) | |
| Neutrophilic dermatoses | |
| Rheumatoid neutrophilic dermatitis (rheumatoid neutrophilic dermatosis) |
|
| Sweet syndrome and pyoderma gangrenosum (PG) * |
|
| Neutrophilic lobular panniculitis |
|
| Vasculitis and vascular reactions | |
| Bywaters lesions |
|
| Rheumatoid vasculitis |
|
| Intravascular/intralymphatic histiocytosis |
|
| Erythema elevatum diutinum |
|
| Complications of therapy for RA | |
| NSAIDs |
|
| Methotrexate |
|
| Methotrexate ≫ TNF inhibitors |
|
| TNF inhibitors |
|
* “PG-like” leg ulcers are a feature of Felty syndrome, which is characterized by the triad of neutropenia, splenomegaly, and RA; Felty syndrome may be associated with T-cell large granular lymphocyte leukemia.
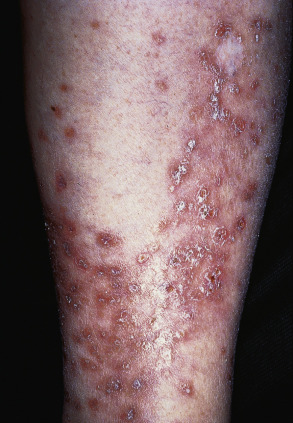
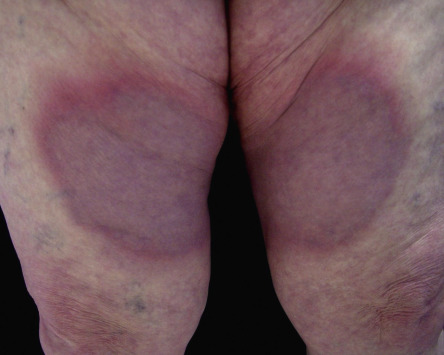
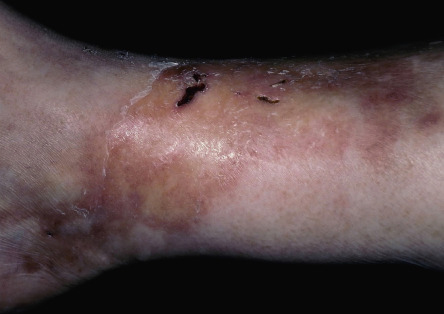
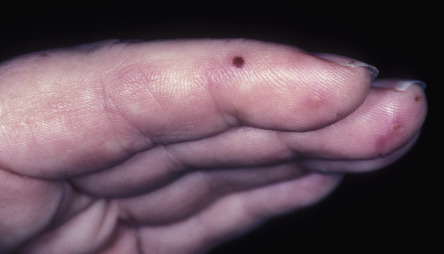
Sjögren syndrome is an autoimmune disorder that is characterized by keratoconjunctivitis sicca and xerostomia (see Table 45.4 ). In primary Sjögren syndrome, vascular inflammation within the skin (e.g. small vessel vasculitis, hypergammaglobulinemic purpura) can occur even before the clinical diagnosis of Sjögren syndrome has been established. Some patients from the Far East, including Japan and the Philippines, have been described with “annular erythema of Sjögren syndrome”. There is ongoing debate as to whether this disorder is unique to Sjögren syndrome or if it is, in fact, a form of subacute cutaneous lupus erythematosus . Secondary Sjögren syndrome occurs in patients with other rheumatologic diseases, including rheumatoid arthritis, dermatomyositis and systemic sclerosis. It is important to remember that the diagnosis of Sjögren syndrome is one of exclusion, and that other conditions can cause xerostomia/xerophthalmia, including chronic GVHD, sarcoidosis, primary systemic amyloidosis, hepatitis C viral infection, and HIV disease.
Cutaneous Hematology and Oncology
Disorders of mutual interest to the dermatologist and hematologist/oncologist are well covered in the following chapters of this textbook: metastatic disease ( Ch. 122 ), leukemia/lymphoma ( Chapter 119 , Chapter 120 , Chapter 121 ), dysproteinemias ( Ch. 119 ), histiocytoses ( Ch. 91 ), vascular neoplasms ( Ch. 114 ), cutaneous T-cell lymphoma ( Ch. 120 ), melanoma ( Ch. 113 ), and cutaneous reactions to chemotherapeutic agents ( Ch. 21 ). Skin signs of internal malignancy are summarized in Tables 53.2 and 53.3 .
| PARANEOPLASTIC DERMATOSES | ||
|---|---|---|
| Dermatoses | Cutaneous findings | Comments |
| Disorders that are associated with cancer in most or all cases | ||
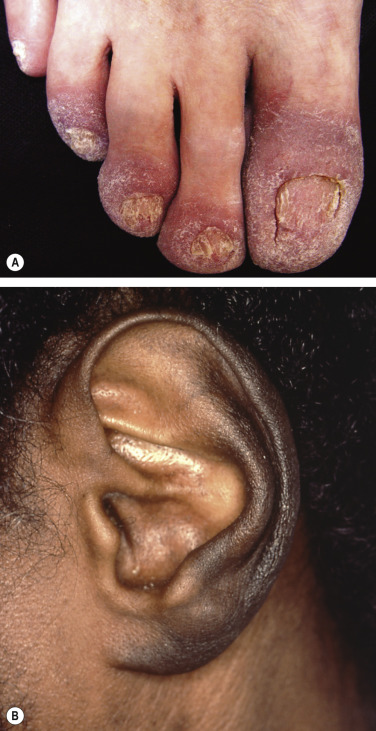
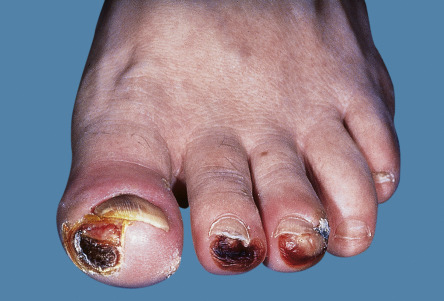
| Bazex syndrome (acrokeratosis paraneoplastica) | Acral psoriasiform plaques, typically with involvement of the nose and helices; often the lesions are violaceous ( Fig. 53.6 ). Longitudinal and horizontal ridging of the nails occurs in 75% of patients | By definition, this condition is linked to malignancy, occurring most commonly in the upper aerodigestive tract (pharynx, larynx or esophagus) |
| Carcinoid syndrome | Flushing and erythema of the head and neck. Pellagra-like dermatitis and sclerodermoid changes may develop in advanced disease | Flushing associated with ~10% of mid-gut tumors (small intestine, appendix, proximal colon) and liver metastases are required; type III gastric and bronchial carcinoid tumors are also associated with flushing (liver metastases are not required) |
| Erythema gyratum repens | Concentric erythematous lesions, often giving rise to a wood-grain appearance ( Fig. 53.7 ; see Ch. 19 ) | Variable sites and types of malignancy |
| Acquired hypertrichosis lanuginosa (malignant down) | Growth of fine lanugo hairs in a generalized distribution or localized to the face. With time, these hairs may become coarser (see Ch. 70 ) | Associated with a variety of internal malignancies, most often carcinoma of the lung, colon or breast |
| AESOP syndrome | Large red to violet–brown patch | Patch overlies a plasmacytoma |
| Ectopic adrenocorticotropic hormone (ACTH) syndrome | Generalized hyperpigmentation that may be accentuated in sun-exposed sites | Production of ACTH by a tumor (often a small cell carcinoma of the lung) may result in hyperpigmentation and features of Cushing syndrome |
| Glucagonoma syndrome | Necrolytic migratory erythema, angular cheilitis, glossitis | Due to a glucagon-secreting tumor of the pancreas. Patients are often treated for intertrigo before the syndrome is diagnosed. Weight loss and diabetes mellitus accompany the dermatosis |
| Paraneoplastic pemphigus | Erosive disease of the mucous membranes and erythema multiforme-like, bullous pemphigoid-like or lichenoid skin lesions ( Fig. 53.8 ; see Ch. 29 ) | Most often associated with non-Hodgkin lymphoma, chronic lymphocytic leukemia or Castleman disease (with the latter accounting for the majority of cases in children and Asian populations). Castleman tumors have been shown to produce the autoantibodies responsible for paraneoplastic pemphigus, and their resection can lead to remission of mucocutaneous lesions. Bronchiolitis obliterans is a common complication |
| Tripe palms | Ridged velvety lesions on the palms | May or may not be accompanied by acanthosis nigricans |
| Disorders that are strongly associated with cancer in a subset of cases | ||
| Acanthosis nigricans | Rapid onset of hyperpigmented velvety changes in multiple flexural sites (e.g. neck, axillae, groin). May also involve extensor surfaces (e.g. elbows, knees, knuckles) and, in malignancy-associated cases, lips, oral mucosa, and palms (see above, tripe palms). Glossitis is also frequently present in patients with malignancy-associated acanthosis nigricans | Association with adenocarcinoma of the stomach or other sites within the GI or GU tracts; in this setting, acanthosis nigricans is often accompanied by weight loss. Acanthosis nigricans is more commonly associated with endocrinologic abnormalities, in particular insulin resistance (see Fig. 53.14 ); such patients are typically overweight, and the onset of the condition is usually insidious |
| Anti-epiligrin cicatricial pemphigoid * /anti-laminin 332 mucous membrane pemphigoid | Oral ulcerations, conjunctival erosions, and scarring. Tense blisters and erosions of the skin may also develop (see Ch. 30 ) | Roughly one-third of patients have or develop cancer within the first year following diagnosis. The cancer is usually an adenocarcinoma and is often at an advanced stage at the time of diagnosis, possibly accounting for the high mortality rate |
| Dermatomyositis (adult) | Heliotrope, Gottron papules, photodistributed poikiloderma, nail-fold overgrowth with dilated capillary loops, pruritus, and diffuse scaling of the scalp (see Ch. 42 ) | Population-based studies demonstrate an over-representation of ovarian, lung, colorectal, and pancreatic carcinomas and non-Hodgkin lymphoma in Caucasians |
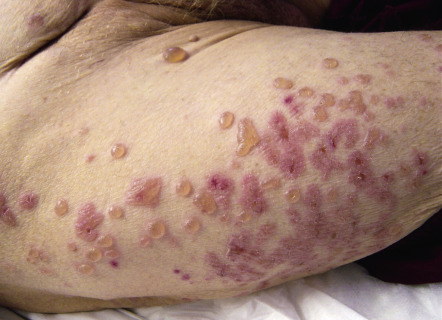
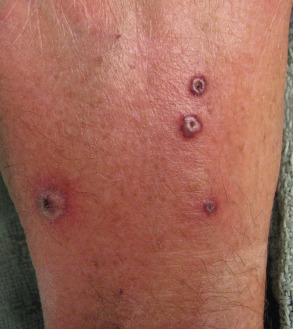
| Neutrophilic dermatoses | Sweet syndrome ( Fig. 53.9 ) or pyoderma gangrenosum (particularly the atypical bullous form) (see Ch. 26 ) | Approximately 10–20% of cases are associated with hematologic disorders such as acute myelogenous leukemia, myelodysplasia or plasma cell dyscrasia (IgA). Underlying solid tumors are rare |
| Disorders associated with a monoclonal gammopathy ** | ||
| Acquired angioedema due to C1 esterase inhibitor dysfunction | Acquired angioedema without associated wheals (see Ch. 18 ) | Associated with B-cell lymphoproliferative disorders, including lymphomas, as well as monoclonal gammopathy of undetermined significance (MGUS) |
| Amyloidosis, primary systemic | Waxy, translucent or purpuric papules; periorbital and pinch purpura; macroglossia (see Ch. 47 ) | Monoclonal gammopathy due to plasma cell dyscrasia ≫ multiple myeloma; deposits composed of immunoglobulin light chain (AL) |
| Cryoglobulinemia, type I | Retiform purpura and necrosis that favors cooler acral sites; acral cyanosis; livedo reticularis (see Ch. 23 ) | Monoclonal gammopathy due to lymphoplasmacytic disorders |
| Necrobiotic xanthogranuloma | Indurated xanthomatous plaques with necrosis and ulceration, usually in a periorbital location (see Ch. 91 ) | Paraproteinemia (>80% of cases, most often IgG with κ light chains); multiple myeloma or a lymphoproliferative disorder develop in a minority of patients |
| Normolipemic plane xanthoma | Yellowish patches and thin plaques that favor the skin folds, upper trunk and periorbital area (see Ch. 92 ) | Paraproteinemia, due to a plasma cell dyscrasia or lymphoproliferative disorder |
| POEMS syndrome ( p olyneuropathy, o rganomegaly, e ndocrinopathy, M -protein and s kin changes) | Although the glomeruloid hemangioma ( Fig. 53.10 ) is considered to be pathognomonic, it is present in a minority of patients. Other skin findings include cherry angiomas, hyperpigmentation, hypertrichosis, sclerodermoid induration, acrocyanosis, acquired facial lipoatrophy, hyperhidrosis, digital clubbing, plethora, and leukonychia | Osteosclerotic myeloma, Castleman disease, and plasmacytomas have been reported in patients with POEMS. In addition to the findings designated in the acronym, patients may have peripheral edema, ascites, pulmonary effusions, papilledema, thrombocytosis, polycythemia, and increased serum levels of VEGF |
| Schnitzler syndrome | Chronic urticarial lesions; histologically, a neutrophil-rich dermal infiltrate is often seen (see Ch. 18 ) | Associated with an IgM, usually κ, paraproteinemia; lymphoplasmacytic malignancies develop in ~15% of patients. Additional manifestations include fevers, arthralgias and bone pain. Entity may be more common than previously reported , and treatment includes anti-IL-1 agents (see Fig. 45.13 ) |
| Scleromyxedema | Sclerodermoid induration plus firm, waxy papules arranged in linear arrays (see Ch. 46 ) | Almost always associated with paraproteinemia (usually IgG with λ light chains); multiple myeloma develops in <10% of cases |
| Dermatoses that may be associated with cancer in a subset of patients | ||
| Acquired ichthyosis | Resembles ichthyosis vulgaris; most often located on the legs | Lymphoma typically predates the diagnosis of the ichthyosis |
| Cutaneous small vessel vasculitis | Palpable purpura; may be widespread and/or in unusual locations (see Ch. 24 ) | Less than 5% of patients with vasculitis have an associated malignancy, most commonly plasma cell dyscrasias, myelodysplasia, myeloproliferative or lymphoproliferative disorders, and hairy cell leukemia |
| Dermatitis herpetiformis (DH) | Pruritic blisters and erosions on extensor surfaces, scalp and/or buttocks (see Ch. 31 ) | Through the association of DH with gluten-sensitive enteropathy, enteropathy-associated T-cell lymphoma occasionally occurs |
| Eruptive disseminated porokeratosis | Abrupt onset of numerous, widespread, inflamed keratoses. Histologically, features of porokeratosis | In a literature review of 35 patients, ~30% of patients had an associated malignancy, most commonly involving the GI tract |
| Exfoliative erythroderma | Widespread, diffuse, scaly erythematous skin (see Ch. 10 ) | May be associated with cutaneous T-cell lymphoma or, occasionally, with a systemic lymphoma or leukemia |
| Juvenile xanthogranulomas (JXGs) in the setting of neurofibromatosis 1 (NF1) | Pink–yellow to red–brown, dome-shaped papules and nodules, most often located on the head and neck (see Ch. 91 ) | A triple association between JXGs, NF1 and JMML has been described |
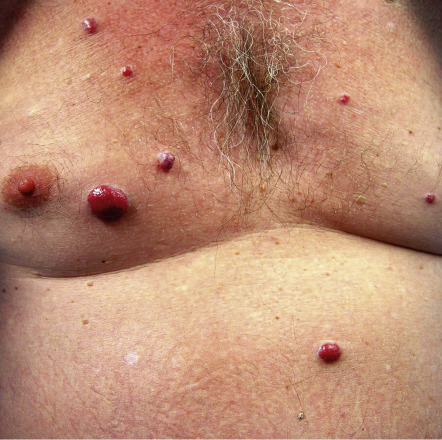
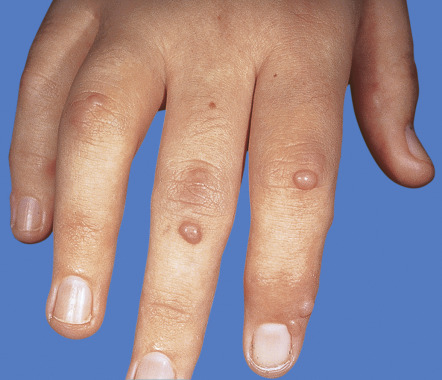
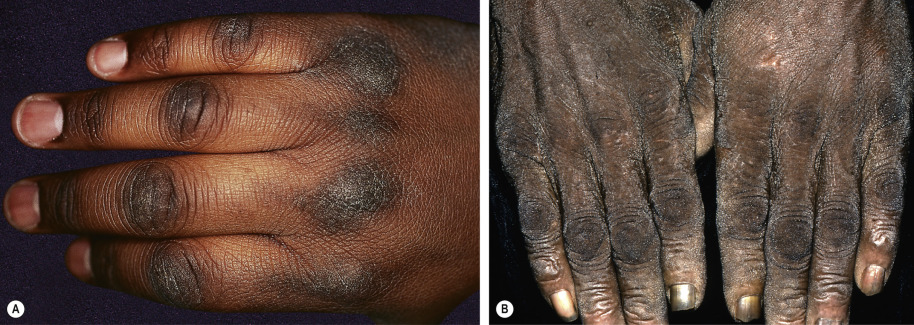
| Multicentric reticulohistiocytosis | Nodular lesions, most often on the dorsal aspects of the hands ( Fig. 53.11 ) | A variety of associated malignancies reported, developing in approximately one-quarter of adult patients |
| Mycosis fungoides | Patch, plaque or nodular disease (see Ch. 120 ) | Some studies have demonstrated an increased risk of second malignancies, especially other lymphoid malignancies |
| Porphyria cutanea tarda (PCT) | Erosions, blisters and scars on the dorsal aspect of the hands, hyperpigmentation, hypertrichosis, milia (see Ch. 49 ) | Through its association with hepatitis C virus, PCT may also be associated with primary hepatocellular carcinoma (hepatoma) |
| Disorders whose association with cancer is controversial | ||
| Sign of Leser–Trélat | Rapid appearance or growth of multiple seborrheic keratoses; keratoses may be inflamed | There is ongoing controversy as to whether this represents a paraneoplastic phenomenon, as many adults have numerous seborrheic keratoses and it can be difficult to determine if the keratoses are truly eruptive. The rapid onset of numerous lesions, particularly if inflamed and associated with diffuse pruritus and/or acanthosis nigricans may warrant evaluation. Eruptive seborrheic keratoses can also develop in erythrodermic patients who do not have an underlying malignancy |
* Evidence for this association is limited by the small number of affected individuals.
** For complete list, see Table 119.4 .
| INTERNAL MALIGNANCIES IN FAMILIAL CANCER SYNDROMES WITH CUTANEOUS MANIFESTATIONS |
|
Both disease-specific and nonspecific skin lesions have been reported in association with myelodysplastic syndrome (MDS) . The former include leukemia cutis and “cutaneous myelodysplasia”; the latter is characterized by an infiltrate of atypical myeloid cells in a patient who does not fulfill the criteria for leukemia. The most commonly associated skin findings include neutrophilic dermatoses and vasculitis. A unique form of Sweet syndrome, which is preceded by a dense perivascular lymphocytic infiltrate, has been described in a few small cohorts of patients with MDS and may portend a poorer prognosis . Generalized granulomatous dermatitis, a variety of autoimmune disorders, including Behçet disease, relapsing polychondritis and bullous pemphigoid, and panniculitis have also been reported. In some patients, these cutaneous disorders precede the diagnosis of MDS, serving as its harbinger. Recurrence of skin lesions, or their lack of response to therapy, may herald progression to leukemia.
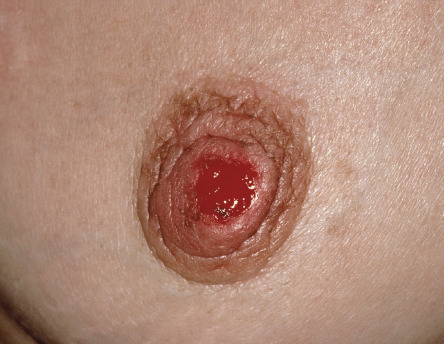
An eczematous or psoriasiform plaque of the nipple and surrounding skin can be seen in Paget disease due to epidermal extension of an underlying ductal adenocarcinoma of the breast ( Fig. 53.12 ). While extramammary Paget disease (EMPD) most commonly represents a primary intraepithelial adenocarcinoma (>75% of patients; Fig. 53.13 ), secondary EMPD can be due to an underlying visceral malignancy, e.g. colon, bladder (see Fig. 73.16 ).
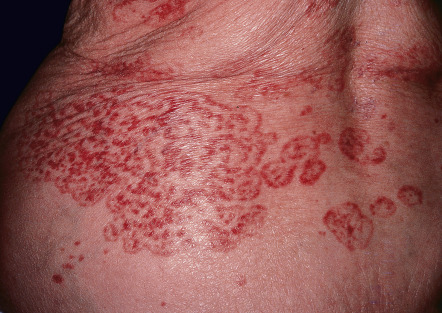
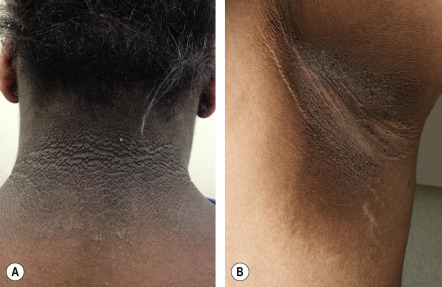
Acanthosis nigricans is characterized by velvety hyperpigmentation of the intertriginous surfaces and, less often, extensor surfaces ( Figs 53.14 & 53.15 ). Although this disorder may be associated with an internal malignancy (most frequently gastric adenocarcinoma), it is most often associated with insulin resistance . The areas most frequently affected are the neck and axillae, but can include any skin fold, including that of the lower lip and chin. Malignancy-associated acanthosis nigricans is usually rapid in onset and may be accompanied by skin tags, multiple seborrheic keratosis, or tripe palms. Verrucous changes of the lips may also develop, and weight loss is common. In contrast, acanthosis nigricans associated with endocrine dysfunction is more insidious in onset and less widespread, and the patients are often overweight or obese. In either instance, there are probably one or more circulating factors that stimulate the epidermal proliferation (i.e. papillomatosis) that is observed histopathologically. An approach to the patient with acanthosis nigricans in outlined in Fig. 53.16 . Treatment of the underlying malignancy, or effective treatment of the endocrinopathy, including weight loss and use of insulin sensitizers, may result in improvement or disappearance of the acanthosis nigricans. Recurrence of malignancy-associated acanthosis nigricans requires a search for recurrent cancer.

The glucagonoma syndrome is characterized by the development of necrolytic migratory erythema (NME), adult-onset diabetes mellitus, weight loss, and glossitis. These patients are also frequently anemic, and personality changes may occur. Skin lesions of NME are eroded, erythematous patches and plaques that characteristically involve the intertriginous areas, face (especially around the mouth), and distal extremities. Blisters may occur and lead to erythematous erosions. The eruption, which can be painful or pruritic, remits and recurs over the course of several weeks to months. It is frequently misdiagnosed as intertrigo or as seborrheic dermatitis. Histologically, pallor of the upper epidermis is highly suggestive of NME or a “nutritional” dermatosis (see Ch. 51 ). However, psoriasiform hyperplasia of the epidermis is often seen in NME and therefore clinicopathologic correlation is required.
Most, but not all, patients with NME have a glucagon-secreting tumor of the pancreas, and effective treatment of the tumor will result in a disappearance of the disease. Rarely, NME develops in the setting of severe liver disease. Similar skin lesions occur in patients with deficiencies of zinc, fatty acids or biotin (see Fig. 51.13 ) and replacement therapy leads to resolution of the cutaneous findings. The differential diagnosis of NME also includes necrolytic acral erythema (see below).
There is growing recognition of the role both inherited and acquired disorders of hypercoagulability or thrombophilia play in various skin disorders (see Chs 23 and 105 ). Some associations, such as those between protein C deficiency and neonatal purpura fulminans or prothrombin gene mutations and livedoid vasculopathy, are fairly well understood. However, there are other disorders in which cutaneous thromboses have been identified (e.g. calciphylaxis) where the precise role remains less well defined. Evaluation for hypercoagulability is warranted in patients with unexplained cutaneous thrombosis (see Table 105.9 ).
Cutaneous Endocrinology and Metabolic Disease
Cutaneous manifestations may provide important clues to the diagnosis of endocrine disorders, including diabetes mellitus . Patients with endocrine diseases are especially susceptible to a number of associated mucocutaneous disorders.
Diabetes mellitus is a very common medical condition that affects almost every organ system. Table 53.4 summarizes cutaneous manifestations of diabetes mellitus ( Figs 53.17–53.22 ). Further details are available in reviews on the subject .
| SELECTED DERMATOLOGIC ASSOCIATIONS OF DIABETES MELLITUS | ||
|---|---|---|
| Dermatosis | Clinical description | Comments |
| Acanthosis nigricans (AN) | Velvety hyperpigmentation of the intertriginous/flexural areas ( Fig. 53.14 ) and, less often, extensor surfaces ( Fig. 53.15A ) | Commonly associated with insulin resistance in both adults and children. Most patients are obese. More common in Hispanics and individuals of African descent. Recent evidence strongly links AN in children with insulin resistance and diabetes |
| Acral dry gangrene | Necrosis of the toes > fingertips | Ischemia due to vascular disease in larger vessels |
| Acral erythema | Erysipelas-like erythema of the hands and/or feet | May be due to small vessel occlusive disease with compensatory hyperemia |
| Carotenoderma | Diffuse orange–yellow skin color | Related to an increase in serum carotene level |
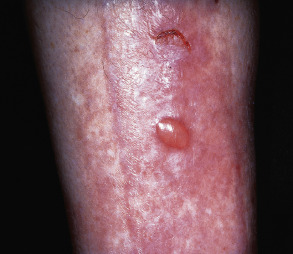
| Diabetic bullae (bullosis diabeticorum) | Tense non-inflammatory bullae on the lower extremities ( Fig. 53.17 ) | Uncertain pathogenesis; microvascular angiopathy possible contributor |
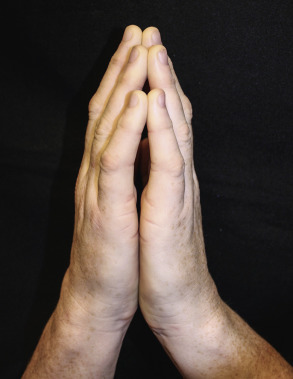
| Diabetic cheiroarthropathy | Thickened skin and limited joint mobility of the hands and fingers, leading to flexion contractures (starting with the fifth digit and progressing radially) and an inability to approximate the palmar surfaces of the hands and fingers (prayer sign; Fig. 53.18 ) | Postulated to result from increased glycosylation of collagen in the skin. Associated with retinopathy, nephropathy, and duration (but not control) of the DM |
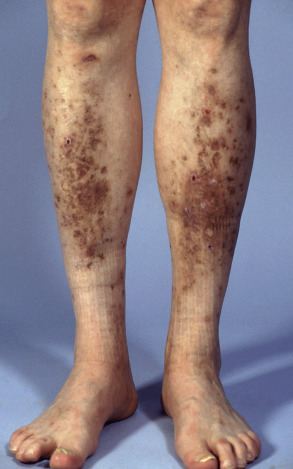
| Diabetic dermopathy | Brown atrophic macules and patches on the legs ( Fig. 53.19 ) | Possibly precipitated by trauma |
| Disseminated granuloma annulare | Erythematous annular lesions composed of papules | There is controversy about the exact relationship of disseminated granuloma annulare and DM (see Ch. 93 ) |
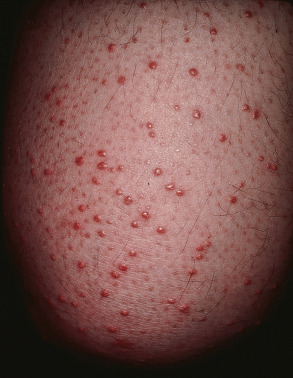
| Eruptive xanthomas | Red–yellow papules that appear over a period of weeks to months ( Fig. 53.20 ) | Associated with elevated serum triglycerides in patients with poorly controlled diabetes. Control of the DM results in resolution of the xanthomas (see Ch. 92 ) |
| Hemochromatosis | Bronzing of the skin due to an increase in melanin rather than iron | Excess of iron stores associated with cirrhosis and cardiac dysfunction as well as DM. Due to mutations in HFE , most commonly C282Y. Also risk factor for porphyria cutanea tarda |
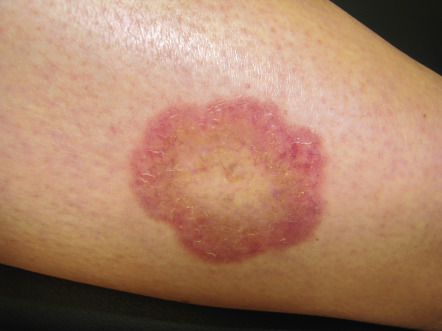
| Necrobiosis lipoidica | Yellow atrophic patches, most often on the shins. A red–brown rim may indicate activity at the border ( Fig. 53.21 ). Ulceration can occur and is often slow to heal | Develops in <1% of patients with DM; diabeticorum was dropped from its name because not always associated with DM (see Ch. 93 ) |
| Neuropathic leg ulcers | Non-painful ulcerations at sites of pressure, most commonly on the foot ( Fig. 53.22 ). When the ulcer is on the plantar surface, a characteristic keratotic rim is seen | Associated with sensory neuropathy (see Ch. 105 ) |
| Perforating disorders (e.g. acquired perforating dermatosis) | Keratotic papules, primarily on the extremities | Often occurs in African-American diabetic patients with chronic kidney disease on dialysis (see Ch. 96 ) |
| Rubeosis | Chronic, flushed appearance of the face, neck and upper extremities | Improved by dietary diabetic control. Flares with vasodilator therapies |
| Scleredema (adultorum of Buschke) | Induration of the upper back and nape due to glycosaminoglycan deposition; may have overlying erythema | No relationship to control of the DM. Also referred to as type III scleredema (see Ch. 46 ) |
The primary characteristics of metabolic syndrome , previously known as syndrome X, are central obesity, hypertension, dyslipidemia, and type 2 diabetes. Almost all patients have insulin resistance, and it is associated with a high risk of cardiovascular disease. Clinical criteria for the diagnosis of metabolic syndrome are outlined in Table 53.5 . Women with polycystic ovary syndrome have a greater frequency of metabolic syndrome and often present with acanthosis nigricans, acne vulgaris, and hirsutism . An association between psoriasis vulgaris and metabolic syndrome has also been identified. Patients with moderate to severe psoriasis are at increased risk of atherosclerotic cardiovascular disease, which is linked to their propensity for the metabolic syndrome. The components of the metabolic syndrome – hyperlipidemia, diabetes mellitus, hypertension and obesity – are all established cardiovascular risk factors .
| CRITERIA FOR THE CLINICAL DIAGNOSIS OF METABOLIC SYNDROME |
|
Thyroid disease is also associated with a significant number of dermatologic disorders. Several reviews have highlighted these dermatologic manifestations and associations , and they are summarized in Table 53.6 .
| DERMATOLOGIC MANIFESTATIONS OF THYROID DISEASE | ||
|---|---|---|
| Hyperthyroidism | Hypothyroidism | |
| Cutaneous changes | Fine, velvety, smooth skin Warm and moist due to increased sweating Hyperpigmentation – localized or generalized Pruritus | Dry, rough, coarse skin Cold and pale Boggy and edematous skin (myxedema) Yellow discoloration as a result of carotenoderma Easy bruising (capillary fragility) |
| Cutaneous diseases | Pretibial myxedema * , thyroid acropachy (acral soft tissue swelling, periostitis, and clubbing of the digits) Urticaria, dermographism Increased incidence of vitiligo | Acquired ichthyosis and palmoplantar keratoderma Eruptive and/or tuberous xanthomas Increased incidence of vitiligo |
| Hair changes/diseases | Fine, thin Mild, diffuse alopecia Increased incidence of alopecia areata | Dull, coarse, brittle Slow growth (increase in telogen hair phase) Alopecia of the lateral third of the eyebrows Increased incidence of alopecia areata |
| Nail changes | Onycholysis | Thin, brittle, striated |
| Koilonychia | Slow growth | |
| Clubbing from thyroid acropachy | Onycholysis (rare) | |
* Can persist when patient is treated and becomes euthyroid or can be associated with euthyroid Graves disease.
Adrenal disease classically manifests as excessive (Cushing syndrome) ( Figs 53.23 & 53.24 ) or insufficient (Addison disease) corticosteroid activity ( Fig. 53.25 ); this can include gluco- and mineralocorticoids. Excessive production of adrenal androgens may also occur (see Ch. 70 ). Features of these syndromes are outlined in Table 53.7 (see Ch. 125 ).
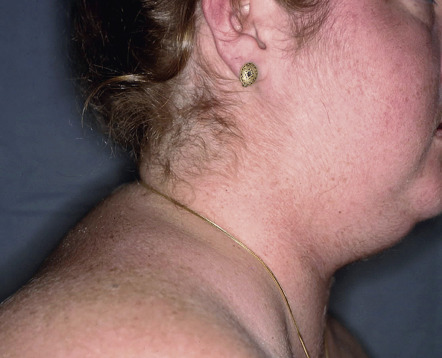
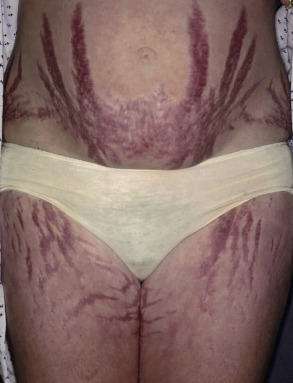
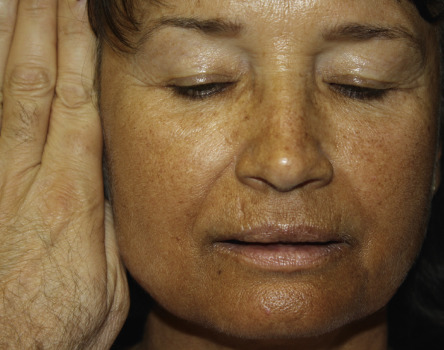
| DERMATOLOGIC MANIFESTATIONS OF ADRENAL DISORDERS | |
|---|---|
| Cushing disease * | Addison disease |
Altered subcutaneous fat distribution
Skin atrophy
Cutaneous infections
Acne Hirsutism | Hyperpigmentation (MSH-like effect of ACTH)
Loss of androgen-stimulated (axillary, pubic) hair in postpubertal women Fibrosis and calcification of cartilage, including ear (rare) In patients with candidiasis endocrinopathy syndrome, vitiligo and chronic mucocutaneous candidiasis |
* Screening tests include late-night salivary cortisol, urine free cortisol (24-hour), 1-mg overnight low-dose dexamethasone suppression test (DST), and longer low-dose DST (2 mg/d for 48 h).
Cutaneous Gastroenterology
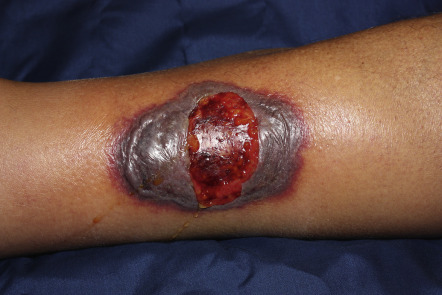
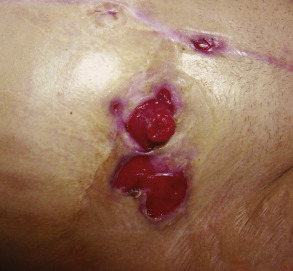
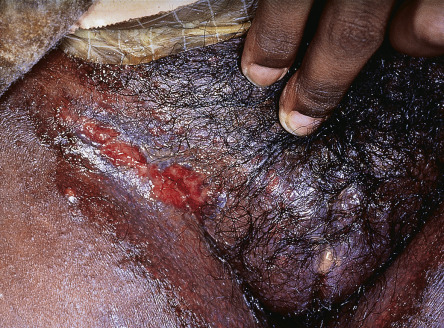
With the exception of Table 26.13 , cutaneous aspects of gastroenterologic or hepatic disease are not specifically discussed elsewhere in this text . Table 53.8 is a list of selected cutaneous associations of Crohn disease and ulcerative colitis ( Figs 53.26–53.28 ). Patients with ostomies, particularly those with an ileostomy, may develop peristomal dermatoses, the most common of which are listed in Table 53.9 . Table 53.10 lists dermatologic conditions associated with gastrointestinal bleeding or hemorrhage ( Figs 53.29–53.32 ) .
| SKIN FINDINGS IN CROHN DISEASE AND ULCERATIVE COLITIS |
|
| PERISTOMAL SKIN DISORDERS | |
|---|---|
| Skin disorder | Comments |
| Irritant contact dermatitis | Most common cause of peristomal dermatitis, especially in patients with an ileostomy. Primarily attributed to exposure to feces or urine |
| Pre-existing skin disease (e.g. psoriasis, seborrheic dermatitis, atopic dermatitis) | Exclude primary contact dermatitis or infection or superimposed contact dermatitis |
| Cutaneous infection ( Candida spp., dermatophyte, herpes virus [primarily simplex], bacteria [especially Staphylococcus aureus ]) | Colonization with bacteria and/or yeast is common. Recent treatment with antibiotics predisposes to candidiasis. May initially improve then worsen if treated inappropriately with topical corticosteroids |
| Allergic contact dermatitis | Relatively uncommon. Potential allergens include: adhesive pastes, adhesive ring or wafer of stoma bag, ostomy bag, epoxy resin, rubber, lanolin, and fragrances. Patch test with both standardized allergens and patient’s own products |
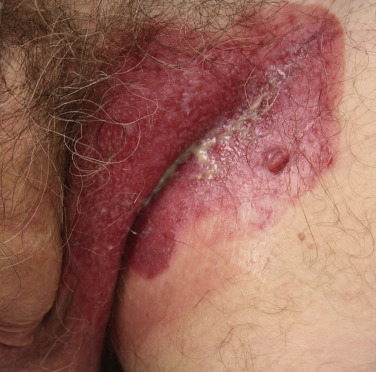
| Pyoderma gangrenosum | Infrequent cause of peristomal dermatitis; most common in patients with inflammatory bowel disease ( Fig. 53.27 ) |
| Pseudo-verrucous papules and nodules | Seen more commonly in association with urostomies; may be misdiagnosed as verrucae |
| CUTANEOUS DISEASES ASSOCIATED WITH GASTROINTESTINAL BLEEDING OR HEMORRHAGE | ||||
|---|---|---|---|---|
| Disorder | Cutaneous findings | Gastrointestinal features | Other pertinent findings | Treatment and comments |
| Genetic (especially those associated with vascular malformations and GI polyps) | ||||
| Hereditary hemorrhagic telangiectasia (HHT) | Macular and papular telangiectasias on facial and acral skin as well as the oral mucosa ( Fig. 53.29 ). Telangiectasias actually represent arteriovenous malformations (AVMs) | Recurrent hemorrhage in the upper GI tract due to AVMs Subset of patients have both juvenile GI polyposis and HHT, most commonly due to mutations in SMAD4 ; associated risk of early-onset colorectal cancer | Epistaxis is often the initial manifestation. AVMs of the lungs, liver and CNS | Autosomal dominant; ~80% of patients have mutations in ENG or ACVRL1 , which encode endoglin and ALK1, TGF-β receptors. Treatment consists of surgical resection, embolization or laser therapy of AVM causing hemorrhage |
| Blue rubber bleb nevus syndrome | Subcutaneous blue to purple venous malformations ( Fig. 53.30 ) | GI vascular malformations | Possible vascular malformations in other organs (e.g. CNS, lungs) | Mosaic due to double (cis) somatic TEK mutations in lesional tissue |
| Pseudoxanthoma elasticum | Yellow papules and plaques on neck and in intertriginous zones, redundant lax skin | Upper or lower GI hemorrhage | Angioid streaks in the eye, hypertension, premature atherosclerosis, uterine hemorrhage, vascular calcification | Autosomal recessive disorder due to mutations in ABCC6 . Diet high in magnesium may be helpful |
| Ehlers–Danlos syndrome, vascular type (type IV) | Translucent fragile skin with extensive bruising | GI hemorrhage due to arterial rupture or intestinal perforation | Minimal joint hyperextensibility (limited to digits), rupture of other arteries or the uterus, acrogeric facies | Autosomal dominant, deficiency of type III collagen |
| Gardner syndrome | Epidermoid cysts, pilomatricomas, lipomas, fibromas, and desmoid tumors | Hemorrhage from adenomatous colonic polyps. Adenocarcinoma of the colon is universal if the colon is not removed | Mandibular and maxillary osteomas and cysts, c ongenital h ypertrophy of the r etinal p igment e pithelium (CHRPE) | Autosomal dominant due to mutations in APC . Colectomy must be appropriately timed |
| Peutz–Jeghers syndrome | Melanotic macules on mucosal surfaces ( Fig. 53.31 ) and, less often, on acral or perioral skin | Hamartomatous polyps throughout the GI tract. Intussusception or hemorrhage may occur | Increased incidence of breast, ovarian sex cord tumors with annular tubules, pancreatic, GI, and other cancers (e.g. lung, endometrial, cervical) | Autosomal dominant due to mutations in STK11 , which encodes a serine/threonine kinase. Remove symptomatic areas of the bowel |
| Cowden syndrome (PTEN hamartoma syndrome; multiple hamartoma syndrome) | Multiple facial or periorificial papules (tricholemmomas), perioral lentigines, cobblestoning of the oral mucosal surface ( Fig. 53.32 ), acral keratotic papules, sclerotic fibromas, lipomas | Hamartomatous polyps throughout the GI tract. Bleeding is rare | Fibrocystic breast disease along with frequent cancers of the breast, often bilateral. Thyroid tumors including cancer | Autosomal dominant due to mutations in the PTEN tumor suppressor gene, which encodes a phosphatase |
| Muir–Torre syndrome | Benign and malignant sebaceous neoplasms and multiple keratoacanthomas | Colorectal and gastric carcinoma > carcinoma of the small bowel | Cancer also may occur in the hepatobiliary and GU tracts | Autosomal dominant due to mutations in one of several mismatch repair genes, including MSH2 (~90%) , MLH1 and MSH6 |
| Inflammatory/autoimmune | ||||
| Cronkhite–Canada syndrome | Circumscribed areas of lentiginous hypermelanosis, alopecia, generalized nail thinning | Adenomatous GI polyps, diarrhea, abdominal pain | Weight loss due to malabsorption | Malignant degeneration is unusual |
| Vasculitis | Palpable purpura, necrotizing livedo reticularis, nodules, or ulcers | Ulcerations secondary to vasculitis of the blood vessels in the bowel. May be more common in IgA-associated vasculitis (Henoch–Schönlein purpura) | Arthritis and nephritis are two additional features of Henoch–Schönlein purpura | Corticosteroids and/or immunosuppressive agents may or may not be effective |
| Malignant atrophic papulosis (Degos disease) | Early lesions – pale red papules with central necrosis Late lesions – atrophic ivory scars | Small infarctions in the GI mucosa. Hemorrhage and intestinal perforation may result in death | CNS involvement, pleuritis, pericarditis | No standard effective therapy. Eculizumab and treprostinil may be beneficial in some patients |
| Ulcerative colitis | Erythema nodosum, vasculitis, pyoderma gangrenosum See Table 53.8 | Uniform and continuous inflammation of the large bowel. Rectal involvement is present in majority (>95%) of patients. Toxic megacolon may develop | Aphthous stomatitis is common. Malignant degeneration in the large intestine is frequent in patients with uncontrolled, longstanding disease | Treatment options include sulfonamides, immunosuppressives, corticosteroids, and total colectomy |
| Crohn disease | Erythema nodosum, vasculitis, pyoderma gangrenosum, perianal fistulae, and metastatic Crohn disease See Table 53.8 | Chronic inflammation, often granulomatous, of the bowel. Skip areas occur | Aphthous stomatitis, arthritis, chronic blood loss | Treatment options include sulfonamides, metronidazole, corticosteroids and other immunosuppressives, including TNF-α inhibitors |
| Nutritional | ||||
| Scurvy | Perifollicular purpura, corkscrew hairs | Collagen degeneration in the vasculature due to vitamin C deficiency. GI blood loss results in anemia | Gingivitis with friable gums and easy bleeding, conjunctival hemorrhage, difficulty walking | Vitamin C replacement |
| Neoplastic | ||||
| Kaposi sarcoma | Violaceous macules, papules, and nodules of the skin and oral mucosa | Similar tumors may occur in the GI tract; possible complications include ulceration, bleeding, perforation and ileus, which are more common in HIV-associated than in classic type | Includes HIV-associated, African-endemic, and classic subtypes, with the latter favoring elderly men of Mediterranean heritage; caused by HHV-8 infection | Chemotherapy or radiation therapy may be helpful |
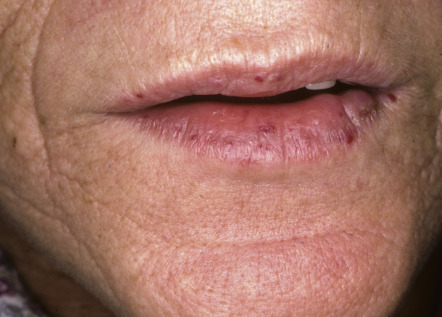
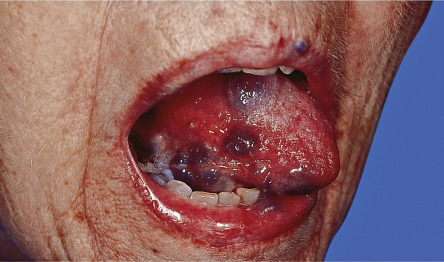
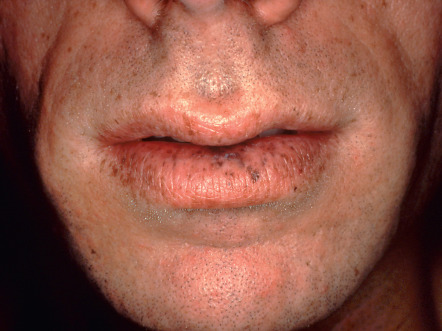
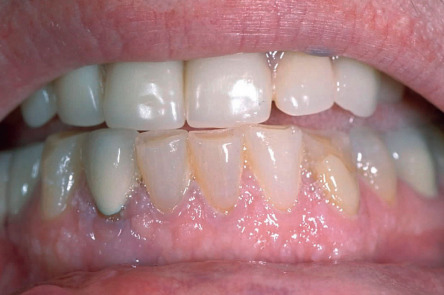
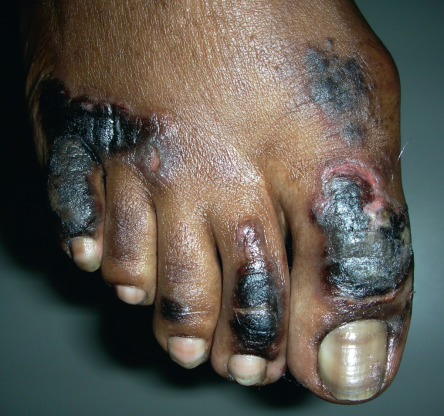
Table 53.11 lists selected dermatologic aspects of liver disease . Necrolytic acral erythema is an interesting disease that has been described in association with hepatitis C viral infection. This entity is histologically similar to necrolytic migratory erythema (see above), but is clinically distinct in that it primarily involves acral sites. The lesions present as painful or pruritic, pink to violet–brown plaques with hyperkeratosis ( Fig. 53.33 ). Blisters and erosions are common, and disease activity tends to wax and wane. Most reported cases have been associated with hepatitis C viral infection, and there has been variable success following treatment with oral zinc supplementation and/or treatment of the underlying hepatitis C viral infection.
| DERMATOLOGIC ASPECTS OF LIVER DISEASE | ||
|---|---|---|
| Liver disease | Dermatologic manifestations/associations | Other features, treatment, and comments |
| Cirrhosis |
|
|
| Primary biliary cirrhosis (PBC) |
| Cholestyramine, rifampin, ursodiol and naloxone may help to relieve pruritus. Osteoporosis is a common complication. Treatment options for PBC include ursodiol, colchicine, methotrexate, and liver transplantation |
| Hemochromatosis (“bronze diabetes”) |
| Due to mutations in HFE , most commonly C282Y. Phlebotomy to reduce iron stores remains primary treatment |
| Wilson disease (hepatolenticular degeneration) |
| Autosomal recessive; due to mutations in ATP7B which encodes a copper-transporting ATPase. Treatment with copper-chelating drugs (penicillamine, trientine) or metallothionein-inducer drugs (zinc acetate); liver transplantation for hepatic failure |
| Hepatitis B and C viruses |
| Treatment of hepatitis viral infections vary based on disease stage, patient comorbidities, and hepatitis C virus genotypes. Options for hepatitis B include: entecavir, tenofovir, lamivudine, adefovir, telbivudine, and interferon. There are multiple treatment regimens for hepatitis C: direct-acting antivirals (DAAs), which include hepatitis C protease and polymerase inhibitors, as well as pegylated interferon + ribavirin |
Other Systemic Diseases With Cutaneous Manifestations
Sarcoidosis is an example of a multisystem disease with a range of cutaneous manifestations (see Ch. 93 ) . Table 53.12 reviews the cardiac and dermatologic features of multisystem disorders ( Figs 53.34 & 53.35 ) . Skin changes associated with pulmonary and renal disorders are summarized in Tables 53.13 and 53.14 , respectively ( Figs 53.36–53.38 ) .
| CARDIOCUTANEOUS ABNORMALITIES IN MULTISYSTEM DISORDERS | |||
|---|---|---|---|
| Disease | Cutaneous features | Cardiovascular findings | Comments |
| Genetic | |||
| Cardio-facio-cutaneous syndrome | Generalized ichthyosis-like scaling, keratosis pilaris, café-au-lait macules, sparse curly hair, sparse or absent eyebrows and eyelashes | Pulmonic stenosis, atrial septal defects, hypertrophic cardiomyopathy | Autosomal dominant “RASopathy” due to mutations in four genes that encode proteins in the RAS/MAPK (mitogen-activated protein kinase) pathway (see Fig. 55.5 ): BRAF > MAP2K1, MAP2K2 > KRAS. Patients have characteristic facies, short stature and developmental delay |
| Costello syndrome | Lax skin on the hands and feet, deep palmoplantar creases, periorificial papillomas, acanthosis nigricans, curly hair | Pulmonic stenosis, hypertrophic cardiomyopathy, arrhythmias | Autosomal dominant “RASopathy” due to mutations in two genes that encode proteins in the RAS/MAPK pathway: HRAS > KRAS . Patients have mental and growth retardation and an increased risk of rhabdomyosarcoma and bladder carcinoma |
| Carvajal syndrome | Striate epidermolytic palmoplantar keratoderma, woolly scalp hair | Dilated left ventricular cardiomyopathy | Autosomal recessive disorder due to desmoplakin mutations |
| Cutis laxa | Lax redundant skin with reduced elasticity | Aortic dilation and rupture, pulmonary artery stenosis, right-sided heart failure | Autosomal dominant (AD) and recessive (AR) forms due to mutations in several genes including those that encode elastin, EFEMP2/fibulin-4, fibulin-5, and V-type H + ATPase subunit (see Table 95.5 ); systemic involvement is more common in the AR form |
| Ehlers–Danlos syndrome (EDS) | Variable degree of skin hyperelasticity and fragility (e.g. “cigarette” paper scars, ecchymoses) | Classic and hypermobility types (I–III): aortic root dilation, mitral and tricuspid valve regurgitation or prolapse | Due to mutations in the genes encoding collagen V (classic type), tenascin X (subset of classic and hypermobility types), collagen III (vascular type), and collagen I (cardiac valvular type) |
| Vascular type: arterial aneurysms, dissection and rupture | |||
| Cardiac valvular type: valvular abnormalities | |||
| Fabry disease | Angiokeratoma corporis diffusum, hypohidrosis | Hypertrophic cardiomyopathy, valvular abnormalities, conduction defects, progressive atherosclerotic disease of the coronary and cerebral arteries | α-Galactosidase A deficiency, X-linked recessive disorder. Cardiac and cerebrovascular disease and renal failure are the usual causes of death |
| Hemochromatosis | Generalized bronze hyperpigmentation | Congestive heart failure, supraventricular arrhythmias | Diabetes, cirrhosis. Genetic testing is available for HFE gene mutations |
| Homocystinuria | Livedo reticularis, malar rash, tissue-paper scars, diffuse pigmentary dilution | Atherosclerosis, arterial and venous thrombosis | Autosomal recessive disorder most commonly due to homozygous cystathionine β-synthase (CBS) deficiency. Rarely due to deficiency of methylenetetrahydrofolate reductase (MTHFR) or other enzymes listed in Table 97.1 . Patients also exhibit marfanoid characteristics and ectopia lentis. Hyperhomocysteinemia (due to homozygous MTHFR deficiency ≫ heterozygous CBS deficiency) also increases risk of arterial and venous thrombosis |
| LEOPARD syndrome/Noonan syndrome with multiple lentigines | Multiple lentigines, café-au-lait and café-noir macules | ECG abnormalities, pulmonary artery stenosis | L – lentigines, E – ECG abnormality, O – ocular hypertelorism, P – pulmonary stenosis, A – abnormalities of the genitalia, R – retardation of growth, D – deafness. |
| Autosomal dominant “RASopathy” due to mutations in PTPN11 (see Fig. 55.5 ) | |||
| Loeys–Dietz syndrome (types I and II) | Translucent skin, easy bruising, atrophic scarring | Aortic aneurysms, arterial tortuosity; significant risk of aortic dissection and rupture, even at an early age | Autosomal dominant disorder due to mutations in the genes that encode TGF-β receptor 1 and 2. Overlapping clinical features with Marfan syndrome and vascular type EDS. Craniofacial anomalies are seen primarily in type I |
| Marfan syndrome | Striae, decreased subcutaneous fat | Dilation and dissection of the ascending aorta, mitral valve prolapse and regurgitation | Autosomal dominant disorder due to mutations in the gene that encodes fibrillin-1. Characterized by tall stature with long limbs, arachnodactyly, dolichocephaly, myopia and ectopia lentis. In addition to β-blockers, angiotensin II receptor blockers (e.g. losartan), which antagonize TGF-β signaling, can prevent aortic root dilation |
| Multiple endocrine neoplasia (MEN) types 2A (Sipple syndrome) and 2B (multiple mucosal neuroma syndrome) | MEN 2A – notalgia paresthetica, macular or lichen amyloidosis, often with childhood onset | Hypertensive crises due to pheochromocytomas (often bilateral) | Autosomal dominant conditions due to mutations in the RET proto-oncogene. Also manifest with medullary cell carcinoma of the thyroid (MEN 2A and 2B), parathyroid adenomas (MEN 2A), and a marfanoid habitus (MEN 2B) |
| MEN 2B – neuromas of the eyelids, lips, tongue and other mucosal sites | |||
| Carney complex (NAME and LAMB syndromes) | Cutaneous myxomas, blue nevi, lentigines, café-au-lait and café-noir macules | Atrial myxoma | Endocrine neoplasia of the adrenal glands, pituitary gland, and/or testes. Autosomal dominant disorder due to mutations in PRKAR1A ; 2 nd genetic locus on chromosome 2 |
| Naxos disease | Diffuse non-epidermolytic palmoplantar keratoderma, woolly scalp hair | Arrhythmogenic right ventricular cardiomyopathy | Autosomal recessive condition due to mutations in the gene that encodes plakoglobin; same clinical findings rarely caused by desmocollin 2 mutations |
| Neurofibromatosis 1 | Café-au-lait macules, neurofibromas, intertriginous freckling (lentigines) | Hypertension, essential or due to pheochromocytoma or renal artery stenosis | Due to mutations in NF1 , which encodes neurofibromin; the latter interacts with the RAS/MAPK pathway (see Fig. 55.5 ) |
| Hutchinson-Gilford progeria syndrome | Sclerodermoid changes, mottled hyperpigmentation, loss of subcutaneous fat, alopecia | Premature atherosclerosis | Autosomal dominant premature aging syndrome due to mutations in the gene encoding lamins A and C |
| Pseudoxanthoma elasticum | Yellow papules and plaques on neck and in intertriginous areas, redundant lax skin | Premature atherosclerotic vascular disease, aortic aneurysm, hypertension | Upper or lower GI hemorrhage, angioid streaks in the eye, uterine hemorrhage. Autosomal recessive disorder due to mutations in ABCC6 |
| Tuberous sclerosis complex (TSC) | Facial angiofibromas, periungual and ungual fibromas, ash leaf macules, shagreen patch | Cardiac rhabdomyomas, arrhythmias | Renal angiomyolipomas, CNS tumors, mental retardation, seizures. Due to mutations in TSC1 or TSC2 , which encode hamartin and tuberin, respectively |
| Werner syndrome | Premature graying, alopecia, sclerodermoid changes, loss of subcutaneous fat, ankle ulcerations | Premature atherosclerosis | Myocardial infarction is usually responsible for death by the fifth decade. Autosomal recessive disorder due to mutations in RECQL2 , which encodes a helicase. Other features include cataracts and malignancy |
| Inflammatory/autoimmune | |||
| Behçet disease | Oral and genital aphthae, pathergy, pustular vasculitis, pyoderma gangrenosum-like lesions, erythema nodosum-like lesions | Pericarditis, coronary arteritis, myocarditis, arrhythmias, valvular disease; vasculitis of the vasa vasorum, primarily of large blood vessels | Ocular and CNS involvement. Inflammatory bowel disease should be excluded |
| Dermatomyositis | Gottron papules, heliotrope rash, photodistributed poikiloderma | Usually asymptomatic; arrhythmias, conduction defects; pericarditis and congestive heart failure are rare | Cardiac involvement is a poor prognostic sign |
| DRESS/DIHS (drug hypersensitivity with eosinophilia and systemic symptoms/ drug-induced hypersensitivity syndrome) | Widespread morbilliform or urticarial eruption or erythroderma; facial edema often present | ECG changes may simulate acute myocardial infarction; acute myocardial dysfunction and hypotension which may initially appear during taper of corticosteroids | Fever, hepatitis, and peripheral eosinophilia common (see Table 21.9 ). Treatment consists of discontinuation of offending drug and corticosteroids; can follow levels of N-terminal pro-brain natriuretic peptide (NT-proBNP). Long-term consequences include thyroid dysfunction and possible autoimmune disease |
| Kawasaki disease (mucocutaneous lymph node syndrome) | Glossitis, cheilitis, acral erythema, edema and desquamation, polymorphous eruption, conjunctival injection | Coronary arteritis, coronary artery aneurysms | High fever, lymphadenopathy. Treatment with IVIg and aspirin significantly decreases risk of coronary artery disease |
| Neonatal lupus erythematosus (NLE) | Transient, non-scarring annular lesions of LE (SCLE-like) that favor the periorbital region | Congenital heart block (CHB) | Presumed to be due to transplacental passage of autoantibodies, most commonly anti-SSA/Ro, but anti-RNP also detected. May have transient cytopenias and hepatitis. Mothers often have a known AI-CTD (~50%) and in those without a known AI-CTD, a significant percentage will develop one over time. For mothers with anti-SSA/Ro antibodies, there is <1% chance of having a baby with NLE, but once have baby with NLE, chance in subsequent pregnancies is 25% |
| Systemic lupus erythematosus | Malar erythema, photosensitivity, lupus skin lesions. Patients with antiphospholipid antibodies may have necrotizing livedo reticularis, leg ulcers or widespread cutaneous necrosis ( Fig. 53.34 ) | Pericarditis, verrucous endocarditis (Libman–Sacks), coronary artery disease | Anti-cardiolipin antibody may play a role. Treatment of LE with corticosteroids may predispose to coronary artery disease |
| Multicentric reticulohistiocytosis | Erythematous nodules of the hands and, less often, the extremities and the face ( Fig. 53.11 ) | Pericarditis, congestive heart failure, coronary artery disease, cardiomegaly | Deforming arthritis is frequent |
| Psoriasis | Well-demarcated, pink-to-red plaques with silvery scale on the elbows, knees and scalp, around the umbilicus, and in the gluteal cleft | Increased risk of cardiovascular disease, including cerebrovascular and peripheral artery disease. In patients with moderate to severe psoriasis, possible increased risk of death from atherosclerotic cardiovascular disease | High incidence of metabolic syndrome in patients with moderate to severe psoriasis (see Table 53.5 ) |
| Relapsing polychondritis | Beefy, red ears (sparing the lobes); inflammation in other tissues with cartilage (e.g. the nose). Late findings include floppy ears and weakness in other areas where the cartilage was affected ( Fig. 53.35 ) | Aortic insufficiency, dissecting aortic aneurysm | Arthritis, tracheal collapse. Dapsone may be helpful. Corticosteroids and/or other immunosuppressive agents are also used |
| Rheumatic fever | Erythema marginatum, subcutaneous nodules | Pancarditis in the acute phase. Late manifestations include mitral and/or aortic valve dysfunction | Rare in US. Follows pharyngitis due to group A β-hemolytic streptococcal infection. Polyarthritis, chorea, fever |
| Sarcoidosis | Papules, nodules, plaques and/or lesions in scars ( Fig. 53.36 ) | Conduction defects, congestive heart failure | Pulmonary disease, lymphadenopathy, hepatic involvement, hypercalcemia. Cardiac involvement denotes a poor prognosis |
| Systemic sclerosis (scleroderma) | Cutaneous sclerosis, Raynaud phenomenon | Pulmonary artery hypertension, conduction defects, pericarditis, visceral Raynaud phenomenon | Cardiac involvement denotes a poor prognosis. May also have interstitial lung disease |
| Vasculitis | Palpable purpura, nodules, livedo reticularis, ulcerations | Coronary artery vasculitis | Arthritis, GI colic or bleeding. Cardiac involvement is uncommon |
| Neoplastic | |||
| Primary systemic amyloidosis (AL) | Pinch purpura, waxy skin, translucent papules, macroglossia | Restrictive cardiomyopathy, conduction disturbances | Proteinuria, urinary light chains, association with plasma cell dyscrasia ≫ multiple myeloma |
| Carcinoid syndrome | Flushing; pellagra-like dermatitis and sclerodermoid features may occur as late manifestations | Endocardial plaques – tricuspid insufficiency, conduction defects | Flushing associated with ~10% of mid-gut tumors (small intestine, appendix, proximal colon) and liver metastases are required; type III gastric and bronchial carcinoid tumors are also associated with flushing (liver metastases are not required) |
| Infectious | |||
| Endocarditis – bacterial or fungal | Purpura, nail-fold infarction, Janeway lesions, Osler’s nodes | Vegetations and dysfunction of the valves | Fever. May simulate vasculitis or cryoglobulinemia |
| Metabolic/endocrine | |||
| Diabetes mellitus | See Table 53.4 | Coronary artery and peripheral artery disease | Occurs with both type 1 and type 2; insulin resistance may contribute |
| Hyperlipidemia | Xanthomas of multiple types | Coronary artery disease | See Ch. 92 |
| Other | |||
| Exfoliative erythroderma | Exfoliative dermatitis | High-output cardiac failure | The eruption may be due to dermatitis, psoriasis, cutaneous T-cell lymphoma, a drug eruption or other causes |
| PHACE(S) syndrome | Segmental infantile hemangioma, most often of the face and neck | Coarctation of the aorta, atrial and ventricular septal defects, anomalous cervical and cerebral arteries | P – posterior fossa malformations, H – hemangioma, A – arterial anomalies, C – cardiac defects and coarctation of the aorta, E – eye anomalies, S – sternal defects and supraumbilical raphe |
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


